Katarzyna Bąbol-Pokora, Magdalena Wołowiec, Katarzyna Popko, Aleksandra Jaworowska, Yenan T. Bryceson, Bianca Tesi, Jan-Inge Henter, Wojciech Młynarski, Wanda Badowska, Walentyna Balwierz, Katarzyna Drabko, Krzysztof Kałwak, Lucyna Maciejka-Kembłowska, Anna Pieczonka, Grażyna Sobol-Milejska, Sylwia Kołtan, Iwona Malinowska, for the Polish Pediatric Hematology, Oncology Society
{"title":"波兰儿科患者原发性噬血细胞淋巴组织细胞病的分子遗传学多样性","authors":"Katarzyna Bąbol-Pokora, Magdalena Wołowiec, Katarzyna Popko, Aleksandra Jaworowska, Yenan T. Bryceson, Bianca Tesi, Jan-Inge Henter, Wojciech Młynarski, Wanda Badowska, Walentyna Balwierz, Katarzyna Drabko, Krzysztof Kałwak, Lucyna Maciejka-Kembłowska, Anna Pieczonka, Grażyna Sobol-Milejska, Sylwia Kołtan, Iwona Malinowska, for the Polish Pediatric Hematology, Oncology Society","doi":"10.1007/s00005-021-00635-4","DOIUrl":null,"url":null,"abstract":"<div><p>Hemophagocytic lymphohistiocytosis (HLH) is a clinical syndrome of life-threatening inflammation caused by an excessive, prolonged and ineffective immune response. An increasing number of HLH cases is recognized in Poland, but the genetic causes of familial HLH (FHL) have not been reported. We investigated the molecular genetics and associated outcomes of pediatric patients who met HLH criteria. We studied 54 patients with HLH, 36 of whom received genetic studies. Twenty-five patients were subjected to direct sequencing of the <i>PRF1</i>, <i>UNC13D</i>, <i>STX11, XIAP</i> and <i>SH2D1A</i> genes. Additionally, 11 patients were subjected to targeted next-generation sequencing. In our study group, 17 patients (31%) were diagnosed with primary HLH, with bi-allelic FHL variants identified in 13 (36%) patients whereas hemizygous changes were identified in 4 patients with X-linked lymphoproliferative diseases. In addition, one patient was diagnosed with X-linked immunodeficiency with magnesium defect, Epstein–Barr virus infection and neoplasia due to a hemizygous <i>MAGT1</i> variant; another newborn was diagnosed with auto-inflammatory syndrome caused by <i>MVK</i> variants. The majority (65%) of FHL patients carried <i>UNC13D</i> pathogenic variants, whereas <i>PRF1</i> variants occurred in two patients. Novel variants in <i>UNC13D</i>, <i>PRF1</i> and <i>XIAP</i> were detected<i>.</i> Epstein–Barr virus was the most common trigger noted in 23 (65%) of the patients with secondary HLH. In three patients with secondary HLH, heterozygous variants of FHL genes were found. Overall survival for the entire study group was 74% with a median of 3.6 years of follow-up. Our results highlight the diversity of molecular causes of primary HLH in Poland.</p></div>","PeriodicalId":8389,"journal":{"name":"Archivum Immunologiae et Therapiae Experimentalis","volume":"69 1","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2021-10-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://link.springer.com/content/pdf/10.1007/s00005-021-00635-4.pdf","citationCount":"3","resultStr":"{\"title\":\"Molecular Genetics Diversity of Primary Hemophagocytic Lymphohistiocytosis among Polish Pediatric Patients\",\"authors\":\"Katarzyna Bąbol-Pokora, Magdalena Wołowiec, Katarzyna Popko, Aleksandra Jaworowska, Yenan T. Bryceson, Bianca Tesi, Jan-Inge Henter, Wojciech Młynarski, Wanda Badowska, Walentyna Balwierz, Katarzyna Drabko, Krzysztof Kałwak, Lucyna Maciejka-Kembłowska, Anna Pieczonka, Grażyna Sobol-Milejska, Sylwia Kołtan, Iwona Malinowska, for the Polish Pediatric Hematology, Oncology Society\",\"doi\":\"10.1007/s00005-021-00635-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Hemophagocytic lymphohistiocytosis (HLH) is a clinical syndrome of life-threatening inflammation caused by an excessive, prolonged and ineffective immune response. An increasing number of HLH cases is recognized in Poland, but the genetic causes of familial HLH (FHL) have not been reported. We investigated the molecular genetics and associated outcomes of pediatric patients who met HLH criteria. We studied 54 patients with HLH, 36 of whom received genetic studies. Twenty-five patients were subjected to direct sequencing of the <i>PRF1</i>, <i>UNC13D</i>, <i>STX11, XIAP</i> and <i>SH2D1A</i> genes. Additionally, 11 patients were subjected to targeted next-generation sequencing. In our study group, 17 patients (31%) were diagnosed with primary HLH, with bi-allelic FHL variants identified in 13 (36%) patients whereas hemizygous changes were identified in 4 patients with X-linked lymphoproliferative diseases. In addition, one patient was diagnosed with X-linked immunodeficiency with magnesium defect, Epstein–Barr virus infection and neoplasia due to a hemizygous <i>MAGT1</i> variant; another newborn was diagnosed with auto-inflammatory syndrome caused by <i>MVK</i> variants. The majority (65%) of FHL patients carried <i>UNC13D</i> pathogenic variants, whereas <i>PRF1</i> variants occurred in two patients. Novel variants in <i>UNC13D</i>, <i>PRF1</i> and <i>XIAP</i> were detected<i>.</i> Epstein–Barr virus was the most common trigger noted in 23 (65%) of the patients with secondary HLH. In three patients with secondary HLH, heterozygous variants of FHL genes were found. Overall survival for the entire study group was 74% with a median of 3.6 years of follow-up. Our results highlight the diversity of molecular causes of primary HLH in Poland.</p></div>\",\"PeriodicalId\":8389,\"journal\":{\"name\":\"Archivum Immunologiae et Therapiae Experimentalis\",\"volume\":\"69 1\",\"pages\":\"\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2021-10-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://link.springer.com/content/pdf/10.1007/s00005-021-00635-4.pdf\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Archivum Immunologiae et Therapiae Experimentalis\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00005-021-00635-4\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"IMMUNOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Archivum Immunologiae et Therapiae Experimentalis","FirstCategoryId":"3","ListUrlMain":"https://link.springer.com/article/10.1007/s00005-021-00635-4","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
Molecular Genetics Diversity of Primary Hemophagocytic Lymphohistiocytosis among Polish Pediatric Patients
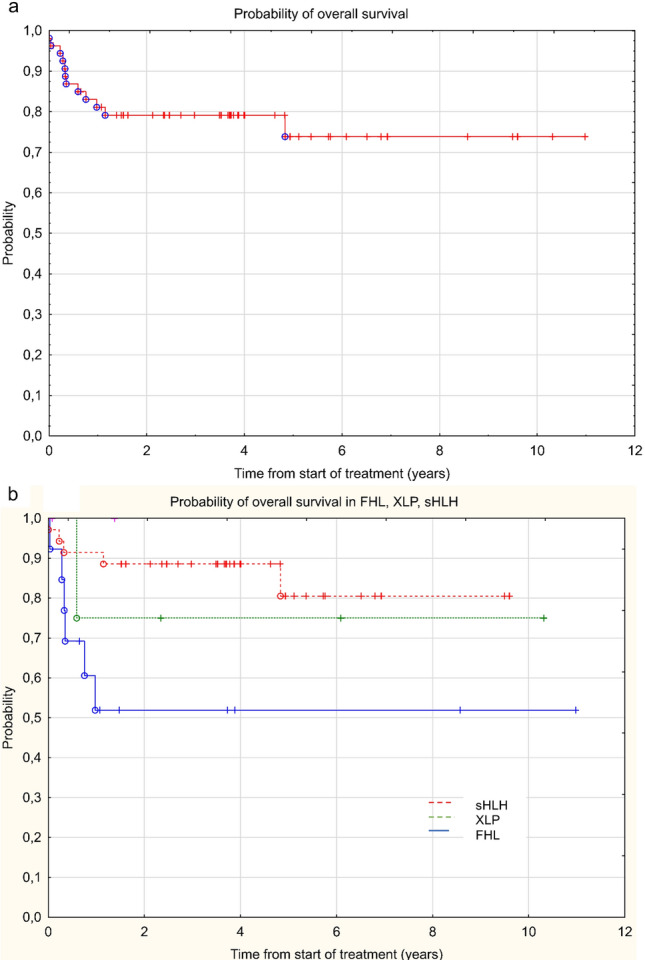
Hemophagocytic lymphohistiocytosis (HLH) is a clinical syndrome of life-threatening inflammation caused by an excessive, prolonged and ineffective immune response. An increasing number of HLH cases is recognized in Poland, but the genetic causes of familial HLH (FHL) have not been reported. We investigated the molecular genetics and associated outcomes of pediatric patients who met HLH criteria. We studied 54 patients with HLH, 36 of whom received genetic studies. Twenty-five patients were subjected to direct sequencing of the PRF1, UNC13D, STX11, XIAP and SH2D1A genes. Additionally, 11 patients were subjected to targeted next-generation sequencing. In our study group, 17 patients (31%) were diagnosed with primary HLH, with bi-allelic FHL variants identified in 13 (36%) patients whereas hemizygous changes were identified in 4 patients with X-linked lymphoproliferative diseases. In addition, one patient was diagnosed with X-linked immunodeficiency with magnesium defect, Epstein–Barr virus infection and neoplasia due to a hemizygous MAGT1 variant; another newborn was diagnosed with auto-inflammatory syndrome caused by MVK variants. The majority (65%) of FHL patients carried UNC13D pathogenic variants, whereas PRF1 variants occurred in two patients. Novel variants in UNC13D, PRF1 and XIAP were detected. Epstein–Barr virus was the most common trigger noted in 23 (65%) of the patients with secondary HLH. In three patients with secondary HLH, heterozygous variants of FHL genes were found. Overall survival for the entire study group was 74% with a median of 3.6 years of follow-up. Our results highlight the diversity of molecular causes of primary HLH in Poland.
期刊介绍:
Archivum Immunologiae et Therapiae Experimentalis (AITE), founded in 1953 by Ludwik Hirszfeld, is a bimonthly, multidisciplinary journal. It publishes reviews and full original papers dealing with immunology, experimental therapy, immunogenetics, transplantation, microbiology, immunochemistry and ethics in science.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
