{"title":"亨廷顿病小鼠模型:揭示CAG重复扩增引起的病理。","authors":"Julia Kaye, Terry Reisine, Steve Finkbeiner","doi":"10.12703/r/10-77","DOIUrl":null,"url":null,"abstract":"<p><p>Huntington's disease (HD) is a neurodegenerative disease that results in motor and cognitive dysfunction, leading to early death. HD is caused by an expansion of CAG repeats in the huntingtin gene (<i>HTT</i>). Here, we review the mouse models of HD. They have been used extensively to better understand the molecular and cellular basis of disease pathogenesis as well as to provide non-human subjects to test the efficacy of potential therapeutics. The first and best-studied <i>in vivo</i> rodent model of HD is the R6/2 mouse, in which a transgene containing the promoter and exon 1 fragment of human <i>HTT</i> with 150 CAG repeats was inserted into the mouse genome. R6/2 mice express rapid, robust behavioral pathologies and display a number of degenerative abnormalities in neuronal populations most vulnerable in HD. The first conditional full-length mutant huntingtin (mHTT) mouse model of HD was the bacterial artificial chromosome (BAC) transgenic mouse model of HD (BACHD), which expresses human full-length m<i>HTT</i> with a mixture of 97 CAG-CAA repeats under the control of endogenous <i>HTT</i> regulatory machinery. It has been useful in identifying the role of mHTT in specific neuronal populations in degenerative processes. In the knock-in (KI) model of HD, the expanded human CAG repeats and human exon 1 are inserted into the mouse <i>Htt</i> locus, so a chimera of the full-length mouse protein with the N-terminal human portion is expressed. Many of aspects of the pathology and behavioral deficits in the KI model better mimic disease characteristics found in HD patients than other models. Accordingly, some have proposed that these mice may be preferable models of the disease over others. Indeed, as our understanding of HD advances, so will the design of animal models to test and develop HD therapies.</p>","PeriodicalId":73016,"journal":{"name":"Faculty reviews","volume":" ","pages":"77"},"PeriodicalIF":0.0000,"publicationDate":"2021-10-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8546598/pdf/","citationCount":"13","resultStr":"{\"title\":\"Huntington's disease mouse models: unraveling the pathology caused by CAG repeat expansion.\",\"authors\":\"Julia Kaye, Terry Reisine, Steve Finkbeiner\",\"doi\":\"10.12703/r/10-77\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Huntington's disease (HD) is a neurodegenerative disease that results in motor and cognitive dysfunction, leading to early death. HD is caused by an expansion of CAG repeats in the huntingtin gene (<i>HTT</i>). Here, we review the mouse models of HD. They have been used extensively to better understand the molecular and cellular basis of disease pathogenesis as well as to provide non-human subjects to test the efficacy of potential therapeutics. The first and best-studied <i>in vivo</i> rodent model of HD is the R6/2 mouse, in which a transgene containing the promoter and exon 1 fragment of human <i>HTT</i> with 150 CAG repeats was inserted into the mouse genome. R6/2 mice express rapid, robust behavioral pathologies and display a number of degenerative abnormalities in neuronal populations most vulnerable in HD. The first conditional full-length mutant huntingtin (mHTT) mouse model of HD was the bacterial artificial chromosome (BAC) transgenic mouse model of HD (BACHD), which expresses human full-length m<i>HTT</i> with a mixture of 97 CAG-CAA repeats under the control of endogenous <i>HTT</i> regulatory machinery. It has been useful in identifying the role of mHTT in specific neuronal populations in degenerative processes. In the knock-in (KI) model of HD, the expanded human CAG repeats and human exon 1 are inserted into the mouse <i>Htt</i> locus, so a chimera of the full-length mouse protein with the N-terminal human portion is expressed. Many of aspects of the pathology and behavioral deficits in the KI model better mimic disease characteristics found in HD patients than other models. Accordingly, some have proposed that these mice may be preferable models of the disease over others. Indeed, as our understanding of HD advances, so will the design of animal models to test and develop HD therapies.</p>\",\"PeriodicalId\":73016,\"journal\":{\"name\":\"Faculty reviews\",\"volume\":\" \",\"pages\":\"77\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2021-10-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8546598/pdf/\",\"citationCount\":\"13\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Faculty reviews\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.12703/r/10-77\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2021/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Faculty reviews","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.12703/r/10-77","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Huntington's disease mouse models: unraveling the pathology caused by CAG repeat expansion.
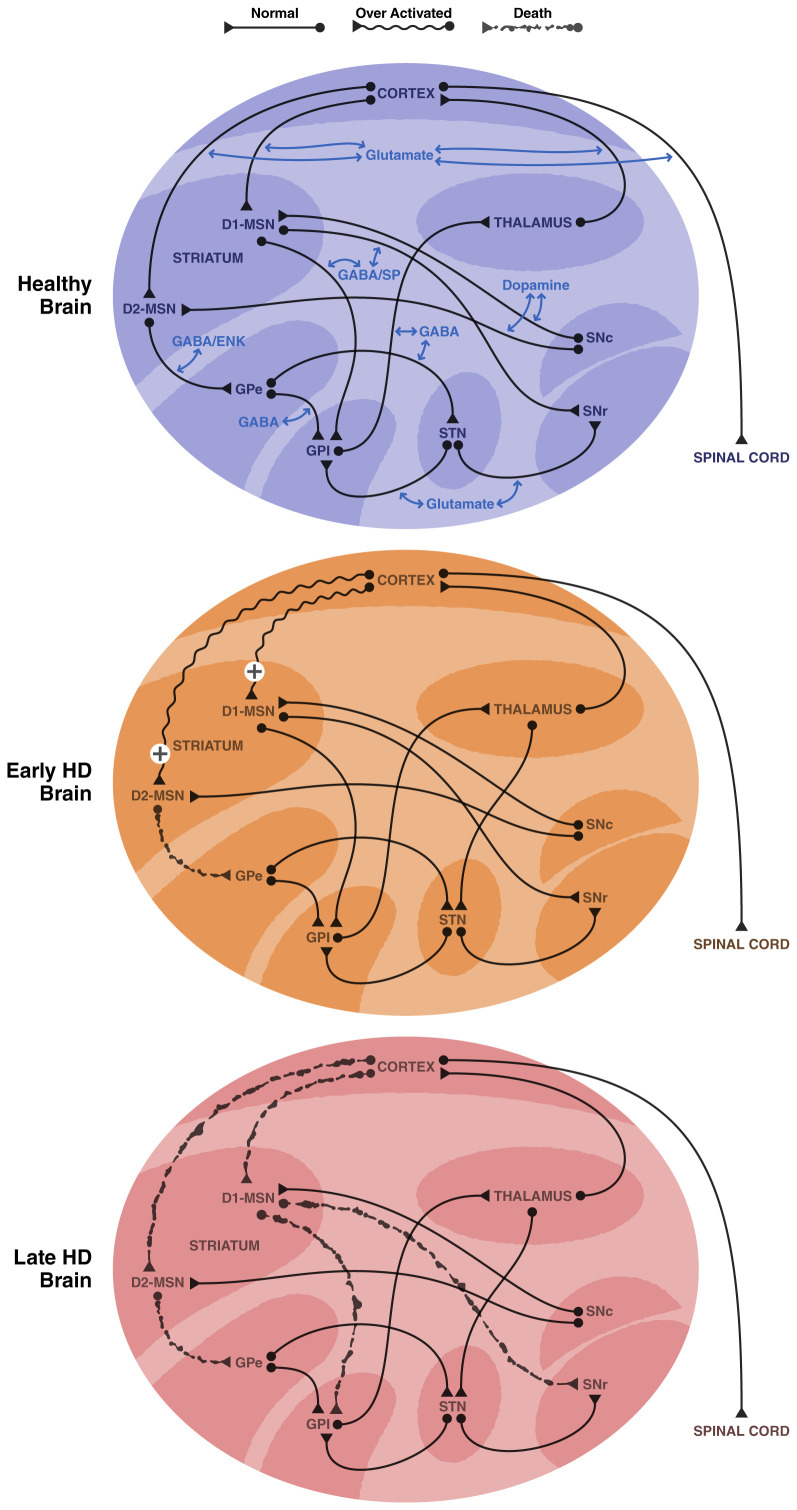
Huntington's disease (HD) is a neurodegenerative disease that results in motor and cognitive dysfunction, leading to early death. HD is caused by an expansion of CAG repeats in the huntingtin gene (HTT). Here, we review the mouse models of HD. They have been used extensively to better understand the molecular and cellular basis of disease pathogenesis as well as to provide non-human subjects to test the efficacy of potential therapeutics. The first and best-studied in vivo rodent model of HD is the R6/2 mouse, in which a transgene containing the promoter and exon 1 fragment of human HTT with 150 CAG repeats was inserted into the mouse genome. R6/2 mice express rapid, robust behavioral pathologies and display a number of degenerative abnormalities in neuronal populations most vulnerable in HD. The first conditional full-length mutant huntingtin (mHTT) mouse model of HD was the bacterial artificial chromosome (BAC) transgenic mouse model of HD (BACHD), which expresses human full-length mHTT with a mixture of 97 CAG-CAA repeats under the control of endogenous HTT regulatory machinery. It has been useful in identifying the role of mHTT in specific neuronal populations in degenerative processes. In the knock-in (KI) model of HD, the expanded human CAG repeats and human exon 1 are inserted into the mouse Htt locus, so a chimera of the full-length mouse protein with the N-terminal human portion is expressed. Many of aspects of the pathology and behavioral deficits in the KI model better mimic disease characteristics found in HD patients than other models. Accordingly, some have proposed that these mice may be preferable models of the disease over others. Indeed, as our understanding of HD advances, so will the design of animal models to test and develop HD therapies.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
