{"title":"基于数据科学的基因组学方法的SARS-CoV-2 rna聚类的高性能计算","authors":"Anas Oujja, Mohamed Riduan Abid, Jaouad Boumhidi, Safae Bourhnane, Asmaa Mourhir, Fatima Merchant, Driss Benhaddou","doi":"10.5808/gi.21056","DOIUrl":null,"url":null,"abstract":"<p><p>Nowadays, Genomic data constitutes one of the fastest growing datasets in the world. As of 2025, it is supposed to become the fourth largest source of Big Data, and thus mandating adequate high-performance computing (HPC) platform for processing. With the latest unprecedented and unpredictable mutations in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the research community is in crucial need for ICT tools to process SARS-CoV-2 RNA data, e.g., by classifying it (i.e., clustering) and thus assisting in tracking virus mutations and predict future ones. In this paper, we are presenting an HPC-based SARS-CoV-2 RNAs clustering tool. We are adopting a data science approach, from data collection, through analysis, to visualization. In the analysis step, we present how our clustering approach leverages on HPC and the longest common subsequence (LCS) algorithm. The approach uses the Hadoop MapReduce programming paradigm and adapts the LCS algorithm in order to efficiently compute the length of the LCS for each pair of SARS-CoV-2 RNA sequences. The latter are extracted from the U.S. National Center for Biotechnology Information (NCBI) Virus repository. The computed LCS lengths are used to measure the dissimilarities between RNA sequences in order to work out existing clusters. In addition to that, we present a comparative study of the LCS algorithm performance based on variable workloads and different numbers of Hadoop worker nodes.</p>","PeriodicalId":36591,"journal":{"name":"Genomics and Informatics","volume":"19 4","pages":"e49"},"PeriodicalIF":0.0000,"publicationDate":"2021-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8752974/pdf/","citationCount":"0","resultStr":"{\"title\":\"High-performance computing for SARS-CoV-2 RNAs clustering: a data science‒based genomics approach.\",\"authors\":\"Anas Oujja, Mohamed Riduan Abid, Jaouad Boumhidi, Safae Bourhnane, Asmaa Mourhir, Fatima Merchant, Driss Benhaddou\",\"doi\":\"10.5808/gi.21056\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Nowadays, Genomic data constitutes one of the fastest growing datasets in the world. As of 2025, it is supposed to become the fourth largest source of Big Data, and thus mandating adequate high-performance computing (HPC) platform for processing. With the latest unprecedented and unpredictable mutations in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the research community is in crucial need for ICT tools to process SARS-CoV-2 RNA data, e.g., by classifying it (i.e., clustering) and thus assisting in tracking virus mutations and predict future ones. In this paper, we are presenting an HPC-based SARS-CoV-2 RNAs clustering tool. We are adopting a data science approach, from data collection, through analysis, to visualization. In the analysis step, we present how our clustering approach leverages on HPC and the longest common subsequence (LCS) algorithm. The approach uses the Hadoop MapReduce programming paradigm and adapts the LCS algorithm in order to efficiently compute the length of the LCS for each pair of SARS-CoV-2 RNA sequences. The latter are extracted from the U.S. National Center for Biotechnology Information (NCBI) Virus repository. The computed LCS lengths are used to measure the dissimilarities between RNA sequences in order to work out existing clusters. In addition to that, we present a comparative study of the LCS algorithm performance based on variable workloads and different numbers of Hadoop worker nodes.</p>\",\"PeriodicalId\":36591,\"journal\":{\"name\":\"Genomics and Informatics\",\"volume\":\"19 4\",\"pages\":\"e49\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2021-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8752974/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genomics and Informatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.5808/gi.21056\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2021/12/31 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"Agricultural and Biological Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genomics and Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5808/gi.21056","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/12/31 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
High-performance computing for SARS-CoV-2 RNAs clustering: a data science‒based genomics approach.
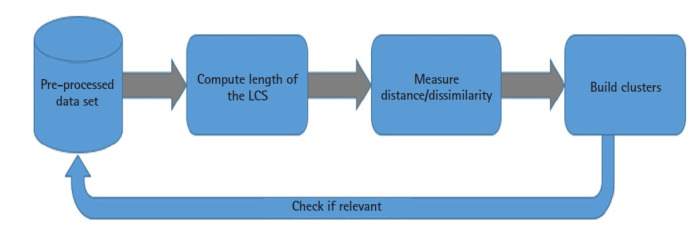
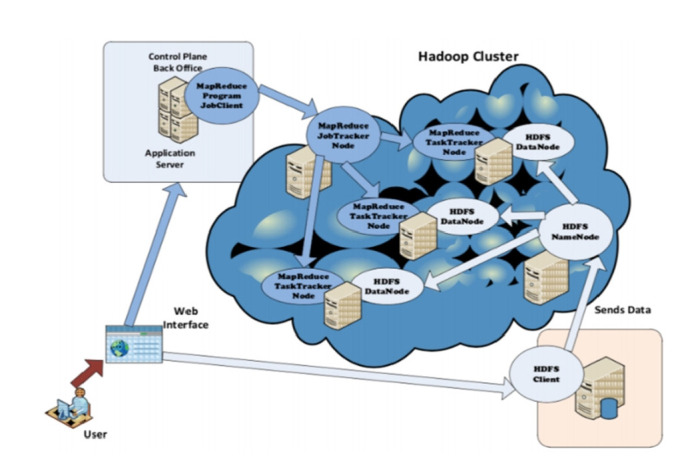
Nowadays, Genomic data constitutes one of the fastest growing datasets in the world. As of 2025, it is supposed to become the fourth largest source of Big Data, and thus mandating adequate high-performance computing (HPC) platform for processing. With the latest unprecedented and unpredictable mutations in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the research community is in crucial need for ICT tools to process SARS-CoV-2 RNA data, e.g., by classifying it (i.e., clustering) and thus assisting in tracking virus mutations and predict future ones. In this paper, we are presenting an HPC-based SARS-CoV-2 RNAs clustering tool. We are adopting a data science approach, from data collection, through analysis, to visualization. In the analysis step, we present how our clustering approach leverages on HPC and the longest common subsequence (LCS) algorithm. The approach uses the Hadoop MapReduce programming paradigm and adapts the LCS algorithm in order to efficiently compute the length of the LCS for each pair of SARS-CoV-2 RNA sequences. The latter are extracted from the U.S. National Center for Biotechnology Information (NCBI) Virus repository. The computed LCS lengths are used to measure the dissimilarities between RNA sequences in order to work out existing clusters. In addition to that, we present a comparative study of the LCS algorithm performance based on variable workloads and different numbers of Hadoop worker nodes.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
