Daniel N Baker, Nathan Dyjack, Vladimir Braverman, Stephanie C Hicks, Ben Langmead
{"title":"快速和高效的scRNA-seq - k-means聚类与不同的距离。","authors":"Daniel N Baker, Nathan Dyjack, Vladimir Braverman, Stephanie C Hicks, Ben Langmead","doi":"10.1145/3459930.3469523","DOIUrl":null,"url":null,"abstract":"Single-cell RNA-sequencing (scRNA-seq) analyses typically begin by clustering a gene-by-cell expression matrix to empirically define groups of cells with similar expression profiles. We describe new methods and a new open source library, minicore, for efficient k-means++ center finding and k-means clustering of scRNA-seq data. Minicore works with sparse count data, as it emerges from typical scRNA-seq experiments, as well as with dense data from after dimensionality reduction. Minicore's novel vectorized weighted reservoir sampling algorithm allows it to find initial k-means++ centers for a 4-million cell dataset in 1.5 minutes using 20 threads. Minicore can cluster using Euclidean distance, but also supports a wider class of measures like Jensen-Shannon Divergence, Kullback-Leibler Divergence, and the Bhattacharyya distance, which can be directly applied to count data and probability distributions. Further, minicore produces lower-cost centerings more efficiently than scikit-learn for scRNA-seq datasets with millions of cells. With careful handling of priors, minicore implements these distance measures with only minor (<2-fold) speed differences among all distances. We show that a minicore pipeline consisting of k-means++, localsearch++ and mini-batch k-means can cluster a 4-million cell dataset in minutes, using less than 10GiB of RAM. This memory-efficiency enables atlas-scale clustering on laptops and other commodity hardware. Finally, we report findings on which distance measures give clusterings that are most consistent with known cell type labels. Availability: The open source library is at https://github.com/dnbaker/minicore. Code used for experiments is at https://github.com/dnbaker/minicore-experiments.","PeriodicalId":72044,"journal":{"name":"ACM-BCB ... ... : the ... ACM Conference on Bioinformatics, Computational Biology and Biomedicine. ACM Conference on Bioinformatics, Computational Biology and Biomedicine","volume":"2021 ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2021-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8586878/pdf/","citationCount":"4","resultStr":"{\"title\":\"Fast and memory-efficient scRNA-seq <i>k</i>-means clustering with various distances.\",\"authors\":\"Daniel N Baker, Nathan Dyjack, Vladimir Braverman, Stephanie C Hicks, Ben Langmead\",\"doi\":\"10.1145/3459930.3469523\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Single-cell RNA-sequencing (scRNA-seq) analyses typically begin by clustering a gene-by-cell expression matrix to empirically define groups of cells with similar expression profiles. We describe new methods and a new open source library, minicore, for efficient k-means++ center finding and k-means clustering of scRNA-seq data. Minicore works with sparse count data, as it emerges from typical scRNA-seq experiments, as well as with dense data from after dimensionality reduction. Minicore's novel vectorized weighted reservoir sampling algorithm allows it to find initial k-means++ centers for a 4-million cell dataset in 1.5 minutes using 20 threads. Minicore can cluster using Euclidean distance, but also supports a wider class of measures like Jensen-Shannon Divergence, Kullback-Leibler Divergence, and the Bhattacharyya distance, which can be directly applied to count data and probability distributions. Further, minicore produces lower-cost centerings more efficiently than scikit-learn for scRNA-seq datasets with millions of cells. With careful handling of priors, minicore implements these distance measures with only minor (<2-fold) speed differences among all distances. We show that a minicore pipeline consisting of k-means++, localsearch++ and mini-batch k-means can cluster a 4-million cell dataset in minutes, using less than 10GiB of RAM. This memory-efficiency enables atlas-scale clustering on laptops and other commodity hardware. Finally, we report findings on which distance measures give clusterings that are most consistent with known cell type labels. Availability: The open source library is at https://github.com/dnbaker/minicore. Code used for experiments is at https://github.com/dnbaker/minicore-experiments.\",\"PeriodicalId\":72044,\"journal\":{\"name\":\"ACM-BCB ... ... : the ... ACM Conference on Bioinformatics, Computational Biology and Biomedicine. ACM Conference on Bioinformatics, Computational Biology and Biomedicine\",\"volume\":\"2021 \",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2021-08-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8586878/pdf/\",\"citationCount\":\"4\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ACM-BCB ... ... : the ... ACM Conference on Bioinformatics, Computational Biology and Biomedicine. ACM Conference on Bioinformatics, Computational Biology and Biomedicine\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1145/3459930.3469523\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACM-BCB ... ... : the ... ACM Conference on Bioinformatics, Computational Biology and Biomedicine. ACM Conference on Bioinformatics, Computational Biology and Biomedicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1145/3459930.3469523","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Fast and memory-efficient scRNA-seq k-means clustering with various distances.
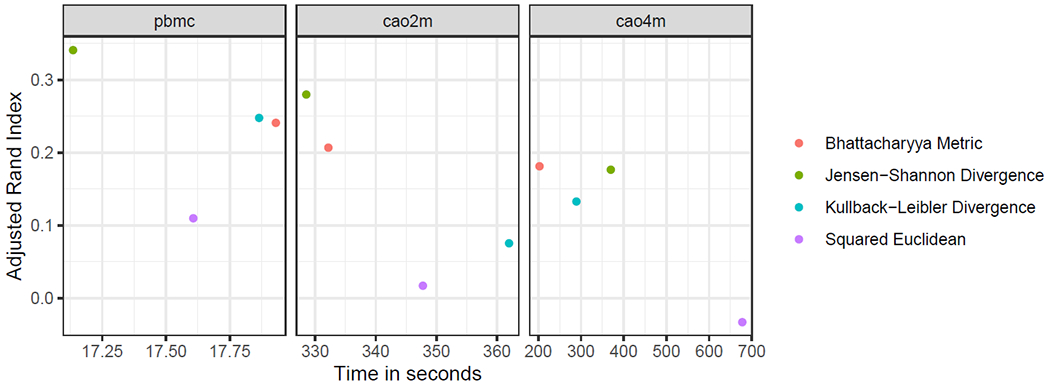
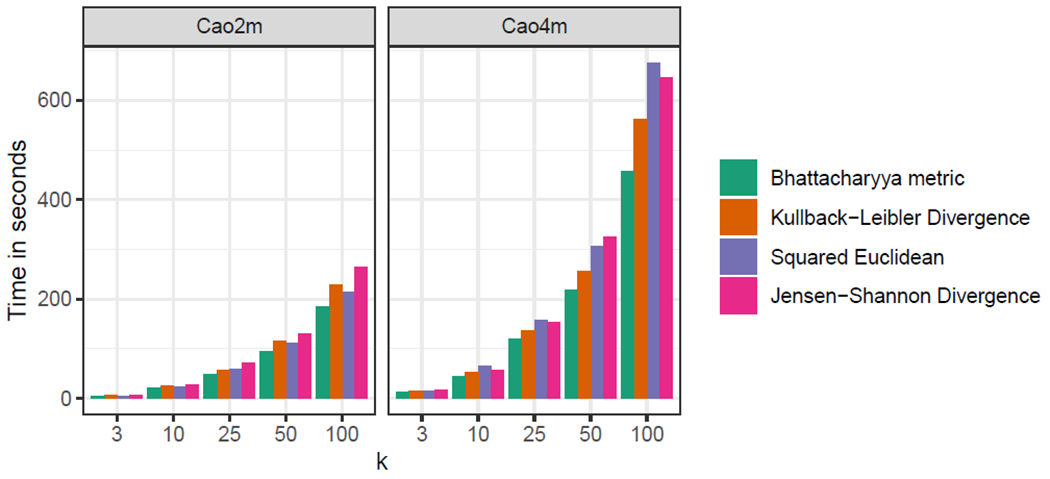
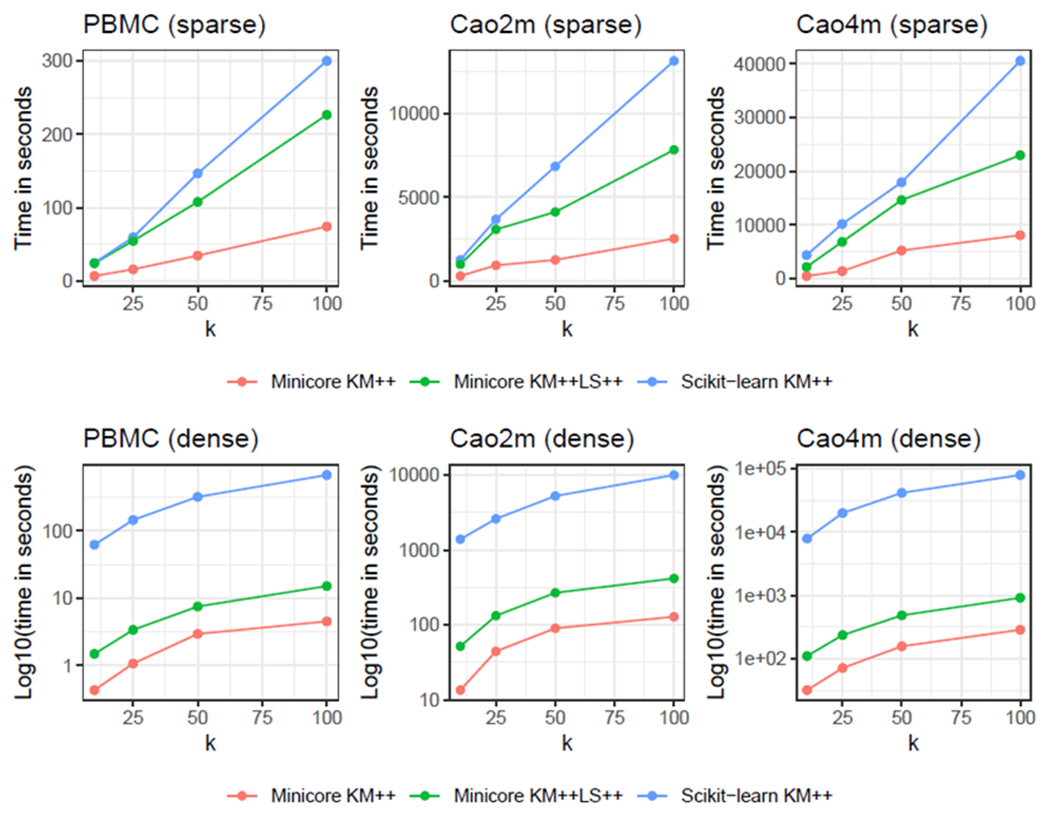
Single-cell RNA-sequencing (scRNA-seq) analyses typically begin by clustering a gene-by-cell expression matrix to empirically define groups of cells with similar expression profiles. We describe new methods and a new open source library, minicore, for efficient k-means++ center finding and k-means clustering of scRNA-seq data. Minicore works with sparse count data, as it emerges from typical scRNA-seq experiments, as well as with dense data from after dimensionality reduction. Minicore's novel vectorized weighted reservoir sampling algorithm allows it to find initial k-means++ centers for a 4-million cell dataset in 1.5 minutes using 20 threads. Minicore can cluster using Euclidean distance, but also supports a wider class of measures like Jensen-Shannon Divergence, Kullback-Leibler Divergence, and the Bhattacharyya distance, which can be directly applied to count data and probability distributions. Further, minicore produces lower-cost centerings more efficiently than scikit-learn for scRNA-seq datasets with millions of cells. With careful handling of priors, minicore implements these distance measures with only minor (<2-fold) speed differences among all distances. We show that a minicore pipeline consisting of k-means++, localsearch++ and mini-batch k-means can cluster a 4-million cell dataset in minutes, using less than 10GiB of RAM. This memory-efficiency enables atlas-scale clustering on laptops and other commodity hardware. Finally, we report findings on which distance measures give clusterings that are most consistent with known cell type labels. Availability: The open source library is at https://github.com/dnbaker/minicore. Code used for experiments is at https://github.com/dnbaker/minicore-experiments.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
