Swetha Ramadesikan, Scott Hickey, Emily De Los Reyes, Anup D Patel, Samuel J Franklin, Patrick Brennan, Erin Crist, Kristy Lee, Peter White, Kim L McBride, Daniel C Koboldt, Richard K Wilson
{"title":"两个患有桥小脑发育不全2D型的兄弟姐妹的双等位SEPSECS变异强调了剪接破坏同义变异在疾病中的相关性。","authors":"Swetha Ramadesikan, Scott Hickey, Emily De Los Reyes, Anup D Patel, Samuel J Franklin, Patrick Brennan, Erin Crist, Kristy Lee, Peter White, Kim L McBride, Daniel C Koboldt, Richard K Wilson","doi":"10.1101/mcs.a006165","DOIUrl":null,"url":null,"abstract":"<p><p>Noncoding and synonymous coding variants that exert their effects via alternative splicing are increasingly recognized as an important category of disease-causing variants. In this report, we describe two siblings who presented with hypotonia, profound developmental delays, and seizures. Brain magnetic resonance imaging (MRI) in the proband at 5 yr showed diffuse cerebral and cerebellar white matter volume loss. Both siblings later developed ventilator-dependent respiratory insufficiency and scoliosis and are currently nonverbal and nonambulatory. Extensive molecular testing including oligo array and clinical exome sequencing was nondiagnostic. Research genome sequencing under an institutional review board (IRB)-approved study protocol revealed that both affected children were compound-heterozygous for variants in the <i>SEPSECS</i> gene. One variant was an initiator codon change (c.1A > T) that disrupted protein translation, consistent with the observation that most disease-causing variants are loss-of-function changes. The other variant was a coding change (c.846G > A) that was predicted to be synonymous but had been demonstrated to disrupt mRNA splicing in a minigene assay. The <i>SEPSECS</i> gene encodes O-phosphoseryl-tRNA(Sec) selenium transferase, an enzyme that participates in the biosynthesis and transport of selenoproteins in the body. Variations in <i>SEPSECS</i> cause autosomal recessive pontocerebellar hypoplasia type 2D (PCHT 2D; OMIM #613811), a neurodegenerative condition characterized by progressive cerebrocerebellar atrophy, microcephaly, and epileptic encephalopathy. The identification of biallelic pathogenic variants in this family-one of which was a synonymous change not identified by prior clinical testing-not only ended the diagnostic odyssey for this family but also highlights the contribution of occult pathogenic variants that may not be recognized by standard genetic testing methodologies.</p>","PeriodicalId":10360,"journal":{"name":"Cold Spring Harbor Molecular Case Studies","volume":" ","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2022-03-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/5d/bf/MCS006165Ram.PMC8958912.pdf","citationCount":"0","resultStr":"{\"title\":\"Biallelic <i>SEPSECS</i> variants in two siblings with pontocerebellar hypoplasia type 2D underscore the relevance of splice-disrupting synonymous variants in disease.\",\"authors\":\"Swetha Ramadesikan, Scott Hickey, Emily De Los Reyes, Anup D Patel, Samuel J Franklin, Patrick Brennan, Erin Crist, Kristy Lee, Peter White, Kim L McBride, Daniel C Koboldt, Richard K Wilson\",\"doi\":\"10.1101/mcs.a006165\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Noncoding and synonymous coding variants that exert their effects via alternative splicing are increasingly recognized as an important category of disease-causing variants. In this report, we describe two siblings who presented with hypotonia, profound developmental delays, and seizures. Brain magnetic resonance imaging (MRI) in the proband at 5 yr showed diffuse cerebral and cerebellar white matter volume loss. Both siblings later developed ventilator-dependent respiratory insufficiency and scoliosis and are currently nonverbal and nonambulatory. Extensive molecular testing including oligo array and clinical exome sequencing was nondiagnostic. Research genome sequencing under an institutional review board (IRB)-approved study protocol revealed that both affected children were compound-heterozygous for variants in the <i>SEPSECS</i> gene. One variant was an initiator codon change (c.1A > T) that disrupted protein translation, consistent with the observation that most disease-causing variants are loss-of-function changes. The other variant was a coding change (c.846G > A) that was predicted to be synonymous but had been demonstrated to disrupt mRNA splicing in a minigene assay. The <i>SEPSECS</i> gene encodes O-phosphoseryl-tRNA(Sec) selenium transferase, an enzyme that participates in the biosynthesis and transport of selenoproteins in the body. Variations in <i>SEPSECS</i> cause autosomal recessive pontocerebellar hypoplasia type 2D (PCHT 2D; OMIM #613811), a neurodegenerative condition characterized by progressive cerebrocerebellar atrophy, microcephaly, and epileptic encephalopathy. The identification of biallelic pathogenic variants in this family-one of which was a synonymous change not identified by prior clinical testing-not only ended the diagnostic odyssey for this family but also highlights the contribution of occult pathogenic variants that may not be recognized by standard genetic testing methodologies.</p>\",\"PeriodicalId\":10360,\"journal\":{\"name\":\"Cold Spring Harbor Molecular Case Studies\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2022-03-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/5d/bf/MCS006165Ram.PMC8958912.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cold Spring Harbor Molecular Case Studies\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1101/mcs.a006165\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/2/1 0:00:00\",\"PubModel\":\"Print\",\"JCR\":\"Q3\",\"JCRName\":\"MEDICINE, RESEARCH & EXPERIMENTAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cold Spring Harbor Molecular Case Studies","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/mcs.a006165","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/2/1 0:00:00","PubModel":"Print","JCR":"Q3","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
Biallelic SEPSECS variants in two siblings with pontocerebellar hypoplasia type 2D underscore the relevance of splice-disrupting synonymous variants in disease.
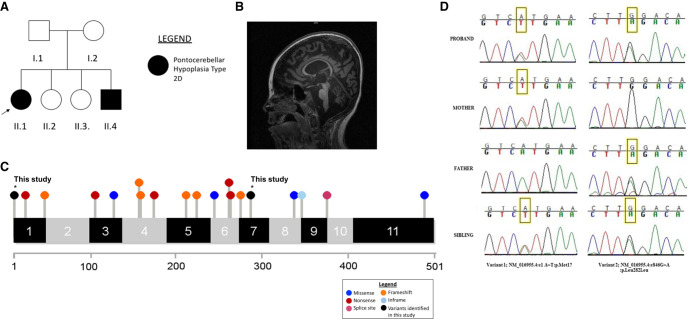
Noncoding and synonymous coding variants that exert their effects via alternative splicing are increasingly recognized as an important category of disease-causing variants. In this report, we describe two siblings who presented with hypotonia, profound developmental delays, and seizures. Brain magnetic resonance imaging (MRI) in the proband at 5 yr showed diffuse cerebral and cerebellar white matter volume loss. Both siblings later developed ventilator-dependent respiratory insufficiency and scoliosis and are currently nonverbal and nonambulatory. Extensive molecular testing including oligo array and clinical exome sequencing was nondiagnostic. Research genome sequencing under an institutional review board (IRB)-approved study protocol revealed that both affected children were compound-heterozygous for variants in the SEPSECS gene. One variant was an initiator codon change (c.1A > T) that disrupted protein translation, consistent with the observation that most disease-causing variants are loss-of-function changes. The other variant was a coding change (c.846G > A) that was predicted to be synonymous but had been demonstrated to disrupt mRNA splicing in a minigene assay. The SEPSECS gene encodes O-phosphoseryl-tRNA(Sec) selenium transferase, an enzyme that participates in the biosynthesis and transport of selenoproteins in the body. Variations in SEPSECS cause autosomal recessive pontocerebellar hypoplasia type 2D (PCHT 2D; OMIM #613811), a neurodegenerative condition characterized by progressive cerebrocerebellar atrophy, microcephaly, and epileptic encephalopathy. The identification of biallelic pathogenic variants in this family-one of which was a synonymous change not identified by prior clinical testing-not only ended the diagnostic odyssey for this family but also highlights the contribution of occult pathogenic variants that may not be recognized by standard genetic testing methodologies.
期刊介绍:
Cold Spring Harbor Molecular Case Studies is an open-access, peer-reviewed, international journal in the field of precision medicine. Articles in the journal present genomic and molecular analyses of individuals or cohorts alongside their clinical presentations and phenotypic information. The journal''s purpose is to rapidly share insights into disease development and treatment gained by application of genomics, proteomics, metabolomics, biomarker analysis, and other approaches. The journal covers the fields of cancer, complex diseases, monogenic disorders, neurological conditions, orphan diseases, infectious disease, gene therapy, and pharmacogenomics. It has a rapid peer-review process that is based on technical evaluation of the analyses performed, not the novelty of findings, and offers a swift, clear path to publication. The journal publishes: Research Reports presenting detailed case studies of individuals and small cohorts, Research Articles describing more extensive work using larger cohorts and/or functional analyses, Rapid Communications presenting the discovery of a novel variant and/or novel phenotype associated with a known disease gene, Rapid Cancer Communications presenting the discovery of a novel variant or combination of variants in a cancer type, Variant Discrepancy Resolution describing efforts to resolve differences or update variant interpretations in ClinVar through case-level data sharing, Follow-up Reports linked to previous observations, Plus Review Articles, Editorials, and Position Statements on best practices for research in precision medicine.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
