{"title":"ACVR1中p.a g258gly伴进行性骨化性纤维发育不良严重变异异位骨化的快速进展:1例病例报告及临床表型回顾","authors":"Kosei Hasegawa, Hiroyuki Tanaka, Natsuko Futagawa, Hiroyuki Miyahara, Hirokazu Tsukahara","doi":"10.1155/2022/5021758","DOIUrl":null,"url":null,"abstract":"<p><p>Fibrodysplasia ossificans progressiva (FOP) is a rare skeletal disorder characterized by congenital malformation of the great toes and progressive heterotopic ossification. Malformation of the great toes appears at birth, while heterotopic ossification generally occurs during childhood and rarely occurs during infancy. Classical FOP results from the heterozygous p.Arg206His variant of the ACVR1 gene, which encodes Activin A receptor type 1. Recently, some atypical FOP patients with other ACVR1 gene variants and clinical features that are not observed in classical FOP patients have been reported. Herein, we describe a girl with severe FOP and multiple anomalies, including syndactyly of the hands and feet, nail agenesis, mandibular hypoplasia, heterotopic ossification occurring from infancy, and congenital cardiac malformation. In our patient, we identified de novo occurrence of the heterozygous p.Arg258Gly variant of ACVR1, which has previously been reported in only two severe FOP patients. Heterotopic ossification occurred earlier and more frequently compared with classical FOP patients. We present the time-series changes in heterotopic ossification in our patient and compare her clinical features with those of the previously reported patients with p.Arg258Gly. Our report deepens understanding of the clinical features in severe FOP with p.Arg258Gly and of FOP as a systemic disorder.</p>","PeriodicalId":30325,"journal":{"name":"Case Reports in Genetics","volume":" ","pages":"5021758"},"PeriodicalIF":0.0000,"publicationDate":"2022-08-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9436604/pdf/","citationCount":"0","resultStr":"{\"title\":\"Rapid Progression of Heterotopic Ossification in Severe Variant of Fibrodysplasia Ossificans Progressiva with p.Arg258Gly in ACVR1: A Case Report and Review of Clinical Phenotypes.\",\"authors\":\"Kosei Hasegawa, Hiroyuki Tanaka, Natsuko Futagawa, Hiroyuki Miyahara, Hirokazu Tsukahara\",\"doi\":\"10.1155/2022/5021758\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Fibrodysplasia ossificans progressiva (FOP) is a rare skeletal disorder characterized by congenital malformation of the great toes and progressive heterotopic ossification. Malformation of the great toes appears at birth, while heterotopic ossification generally occurs during childhood and rarely occurs during infancy. Classical FOP results from the heterozygous p.Arg206His variant of the ACVR1 gene, which encodes Activin A receptor type 1. Recently, some atypical FOP patients with other ACVR1 gene variants and clinical features that are not observed in classical FOP patients have been reported. Herein, we describe a girl with severe FOP and multiple anomalies, including syndactyly of the hands and feet, nail agenesis, mandibular hypoplasia, heterotopic ossification occurring from infancy, and congenital cardiac malformation. In our patient, we identified de novo occurrence of the heterozygous p.Arg258Gly variant of ACVR1, which has previously been reported in only two severe FOP patients. Heterotopic ossification occurred earlier and more frequently compared with classical FOP patients. We present the time-series changes in heterotopic ossification in our patient and compare her clinical features with those of the previously reported patients with p.Arg258Gly. Our report deepens understanding of the clinical features in severe FOP with p.Arg258Gly and of FOP as a systemic disorder.</p>\",\"PeriodicalId\":30325,\"journal\":{\"name\":\"Case Reports in Genetics\",\"volume\":\" \",\"pages\":\"5021758\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2022-08-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9436604/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Case Reports in Genetics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2022/5021758\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2022/5021758","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Rapid Progression of Heterotopic Ossification in Severe Variant of Fibrodysplasia Ossificans Progressiva with p.Arg258Gly in ACVR1: A Case Report and Review of Clinical Phenotypes.
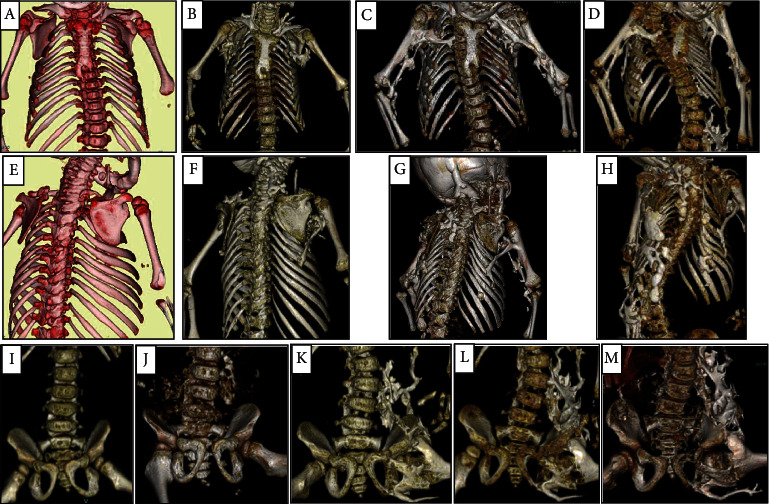
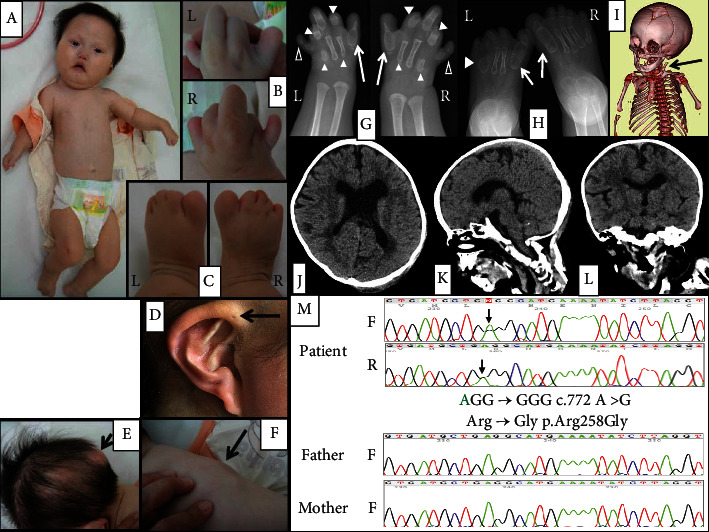
Fibrodysplasia ossificans progressiva (FOP) is a rare skeletal disorder characterized by congenital malformation of the great toes and progressive heterotopic ossification. Malformation of the great toes appears at birth, while heterotopic ossification generally occurs during childhood and rarely occurs during infancy. Classical FOP results from the heterozygous p.Arg206His variant of the ACVR1 gene, which encodes Activin A receptor type 1. Recently, some atypical FOP patients with other ACVR1 gene variants and clinical features that are not observed in classical FOP patients have been reported. Herein, we describe a girl with severe FOP and multiple anomalies, including syndactyly of the hands and feet, nail agenesis, mandibular hypoplasia, heterotopic ossification occurring from infancy, and congenital cardiac malformation. In our patient, we identified de novo occurrence of the heterozygous p.Arg258Gly variant of ACVR1, which has previously been reported in only two severe FOP patients. Heterotopic ossification occurred earlier and more frequently compared with classical FOP patients. We present the time-series changes in heterotopic ossification in our patient and compare her clinical features with those of the previously reported patients with p.Arg258Gly. Our report deepens understanding of the clinical features in severe FOP with p.Arg258Gly and of FOP as a systemic disorder.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
