Fernando Freua, Mariana Espíndola de Castro Almeida, Paulo Ribeiro Nóbrega, Anderson Rodrigues Brandáo de Paiva, Bruno Della-Ripa, Paulina Cunha, Lúcia Inês Macedo-Souza, Clarissa Bueno, David S Lynch, Henry Houlden, Leandro Tavares Lucato, Fernando Kok
{"title":"精氨酸酶 1 缺乏症表现为复杂的遗传性痉挛性截瘫。","authors":"Fernando Freua, Mariana Espíndola de Castro Almeida, Paulo Ribeiro Nóbrega, Anderson Rodrigues Brandáo de Paiva, Bruno Della-Ripa, Paulina Cunha, Lúcia Inês Macedo-Souza, Clarissa Bueno, David S Lynch, Henry Houlden, Leandro Tavares Lucato, Fernando Kok","doi":"10.1101/mcs.a006232","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Argininemia or arginase deficiency is a metabolic disorder caused by pathogenic variants in ARG1 and consists of a variable association of progressive spastic paraplegia, intellectual disability, and seizures. Hereditary spastic paraplegia (HSP) is a group of inherited diseases whose main feature is a progressive gait disorder characterized by lower limb spasticity. This study presents 7 patients with arginase 1 deficiency from 6 different families, all with an initial diagnosis of complicated HSP.</p><p><strong>Methods: </strong>We evaluated the clinical data of 7 patients belonging to six independent families who were diagnosed with hyperargininemia in a neurogenetics outpatient clinic.</p><p><strong>Results: </strong>All patients had lower limb spasticity and six had global developmental delay. Five individuals had intellectual disability and two had epilepsy. Psychiatric abnormalities were seen in two patients. In two participants of this study, MRI disclosed thinning of the corpus callosum. Molecular diagnosis was made by whole exome sequencing. All variants were present in homozygosis; we identified two novel missense variants, one novel frameshift variant, and one previously published missense variant.</p><p><strong>Discussion: </strong>Clinical diagnosis of early onset complicated hereditary spastic paraplegia was made in all patients. Two patients were initially suspected of having SPG11 due to thinning of the corpus callosum. As argininemia may present with a highly penetrant phenotype of spastic paraplegia associated with additional symptoms, this disease may represent a specific entity amongst the complicated HSPs.</p>","PeriodicalId":10360,"journal":{"name":"Cold Spring Harbor Molecular Case Studies","volume":" ","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2022-09-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/09/b3/MCS006232Fre.PMC9632362.pdf","citationCount":"0","resultStr":"{\"title\":\"Arginase 1 deficiency presenting as complicated hereditary spastic paraplegia.\",\"authors\":\"Fernando Freua, Mariana Espíndola de Castro Almeida, Paulo Ribeiro Nóbrega, Anderson Rodrigues Brandáo de Paiva, Bruno Della-Ripa, Paulina Cunha, Lúcia Inês Macedo-Souza, Clarissa Bueno, David S Lynch, Henry Houlden, Leandro Tavares Lucato, Fernando Kok\",\"doi\":\"10.1101/mcs.a006232\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Introduction: </strong>Argininemia or arginase deficiency is a metabolic disorder caused by pathogenic variants in ARG1 and consists of a variable association of progressive spastic paraplegia, intellectual disability, and seizures. Hereditary spastic paraplegia (HSP) is a group of inherited diseases whose main feature is a progressive gait disorder characterized by lower limb spasticity. This study presents 7 patients with arginase 1 deficiency from 6 different families, all with an initial diagnosis of complicated HSP.</p><p><strong>Methods: </strong>We evaluated the clinical data of 7 patients belonging to six independent families who were diagnosed with hyperargininemia in a neurogenetics outpatient clinic.</p><p><strong>Results: </strong>All patients had lower limb spasticity and six had global developmental delay. Five individuals had intellectual disability and two had epilepsy. Psychiatric abnormalities were seen in two patients. In two participants of this study, MRI disclosed thinning of the corpus callosum. Molecular diagnosis was made by whole exome sequencing. All variants were present in homozygosis; we identified two novel missense variants, one novel frameshift variant, and one previously published missense variant.</p><p><strong>Discussion: </strong>Clinical diagnosis of early onset complicated hereditary spastic paraplegia was made in all patients. Two patients were initially suspected of having SPG11 due to thinning of the corpus callosum. As argininemia may present with a highly penetrant phenotype of spastic paraplegia associated with additional symptoms, this disease may represent a specific entity amongst the complicated HSPs.</p>\",\"PeriodicalId\":10360,\"journal\":{\"name\":\"Cold Spring Harbor Molecular Case Studies\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2022-09-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/09/b3/MCS006232Fre.PMC9632362.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cold Spring Harbor Molecular Case Studies\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1101/mcs.a006232\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"MEDICINE, RESEARCH & EXPERIMENTAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cold Spring Harbor Molecular Case Studies","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/mcs.a006232","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
Arginase 1 deficiency presenting as complicated hereditary spastic paraplegia.
Introduction: Argininemia or arginase deficiency is a metabolic disorder caused by pathogenic variants in ARG1 and consists of a variable association of progressive spastic paraplegia, intellectual disability, and seizures. Hereditary spastic paraplegia (HSP) is a group of inherited diseases whose main feature is a progressive gait disorder characterized by lower limb spasticity. This study presents 7 patients with arginase 1 deficiency from 6 different families, all with an initial diagnosis of complicated HSP.
Methods: We evaluated the clinical data of 7 patients belonging to six independent families who were diagnosed with hyperargininemia in a neurogenetics outpatient clinic.
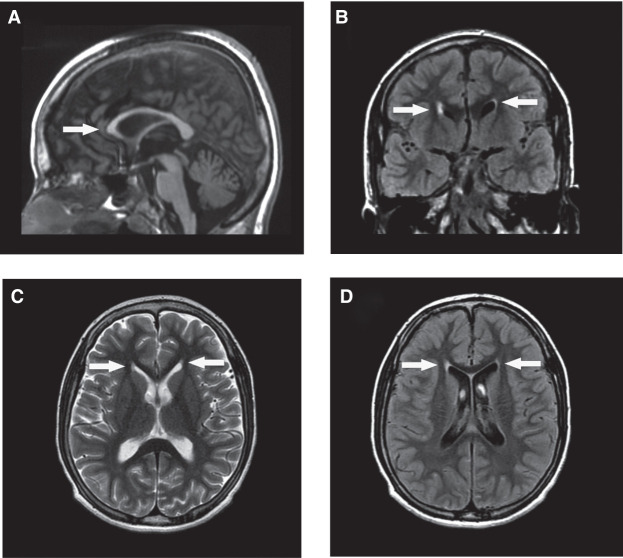
Results: All patients had lower limb spasticity and six had global developmental delay. Five individuals had intellectual disability and two had epilepsy. Psychiatric abnormalities were seen in two patients. In two participants of this study, MRI disclosed thinning of the corpus callosum. Molecular diagnosis was made by whole exome sequencing. All variants were present in homozygosis; we identified two novel missense variants, one novel frameshift variant, and one previously published missense variant.
Discussion: Clinical diagnosis of early onset complicated hereditary spastic paraplegia was made in all patients. Two patients were initially suspected of having SPG11 due to thinning of the corpus callosum. As argininemia may present with a highly penetrant phenotype of spastic paraplegia associated with additional symptoms, this disease may represent a specific entity amongst the complicated HSPs.
期刊介绍:
Cold Spring Harbor Molecular Case Studies is an open-access, peer-reviewed, international journal in the field of precision medicine. Articles in the journal present genomic and molecular analyses of individuals or cohorts alongside their clinical presentations and phenotypic information. The journal''s purpose is to rapidly share insights into disease development and treatment gained by application of genomics, proteomics, metabolomics, biomarker analysis, and other approaches. The journal covers the fields of cancer, complex diseases, monogenic disorders, neurological conditions, orphan diseases, infectious disease, gene therapy, and pharmacogenomics. It has a rapid peer-review process that is based on technical evaluation of the analyses performed, not the novelty of findings, and offers a swift, clear path to publication. The journal publishes: Research Reports presenting detailed case studies of individuals and small cohorts, Research Articles describing more extensive work using larger cohorts and/or functional analyses, Rapid Communications presenting the discovery of a novel variant and/or novel phenotype associated with a known disease gene, Rapid Cancer Communications presenting the discovery of a novel variant or combination of variants in a cancer type, Variant Discrepancy Resolution describing efforts to resolve differences or update variant interpretations in ClinVar through case-level data sharing, Follow-up Reports linked to previous observations, Plus Review Articles, Editorials, and Position Statements on best practices for research in precision medicine.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
