Anna Lengyel, Éva Pinti, Henriett Pikó, Árvai Kristóf, Tünde Abonyi, Zaránd Némethi, György Fekete, Irén Haltrich
{"title":"在匈牙利神经发育障碍患者队列中通过染色体微阵列鉴定的罕见拷贝数变异的临床评价。","authors":"Anna Lengyel, Éva Pinti, Henriett Pikó, Árvai Kristóf, Tünde Abonyi, Zaránd Némethi, György Fekete, Irén Haltrich","doi":"10.1186/s13039-022-00623-z","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Neurodevelopmental disorders are genetically heterogeneous pediatric conditions. The first tier diagnostic method for uncovering copy number variations (CNVs), one of the most common genetic etiologies in affected individuals, is chromosomal microarray (CMA). However, this methodology is not yet a routine molecular cytogenetic test in many parts of the world, including Hungary. Here we report clinical and genetic data of the first, relatively large Hungarian cohort of patients whose genetic testing included CMA.</p><p><strong>Methods: </strong>Clinical data were retrospectively collected for 78 children who were analyzed using various CMA platforms. Phenotypes of patients with disease-causing variants were compared to patients with negative results using the chi squared/Fisher exact tests.</p><p><strong>Results: </strong>A total of 30 pathogenic CNVs were identified in 29 patients (37.2%). Postnatal growth delay (p = 0.05564), pectus excavatum (p = 0.07484), brain imaging abnormalities (p = 0.07848), global developmental delay (p = 0.08070) and macrocephaly (p = 0.08919) were more likely to be associated with disease-causing CNVs.</p><p><strong>Conclusion: </strong>Our results allow phenotypic expansion of 14q11.2 microdeletions encompassing SUPT16H and CHD8 genes. Variants of unknown significance (n = 24) were found in 17 patients. We provide detailed phenotypic and genetic data of these individuals to facilitate future classification efforts, and spotlight two patients with potentially pathogenic alterations. Our results contribute to unraveling the diagnostic value of rare CNVs.</p>","PeriodicalId":19099,"journal":{"name":"Molecular Cytogenetics","volume":null,"pages":null},"PeriodicalIF":1.3000,"publicationDate":"2022-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9623912/pdf/","citationCount":"2","resultStr":"{\"title\":\"Clinical evaluation of rare copy number variations identified by chromosomal microarray in a Hungarian neurodevelopmental disorder patient cohort.\",\"authors\":\"Anna Lengyel, Éva Pinti, Henriett Pikó, Árvai Kristóf, Tünde Abonyi, Zaránd Némethi, György Fekete, Irén Haltrich\",\"doi\":\"10.1186/s13039-022-00623-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Neurodevelopmental disorders are genetically heterogeneous pediatric conditions. The first tier diagnostic method for uncovering copy number variations (CNVs), one of the most common genetic etiologies in affected individuals, is chromosomal microarray (CMA). However, this methodology is not yet a routine molecular cytogenetic test in many parts of the world, including Hungary. Here we report clinical and genetic data of the first, relatively large Hungarian cohort of patients whose genetic testing included CMA.</p><p><strong>Methods: </strong>Clinical data were retrospectively collected for 78 children who were analyzed using various CMA platforms. Phenotypes of patients with disease-causing variants were compared to patients with negative results using the chi squared/Fisher exact tests.</p><p><strong>Results: </strong>A total of 30 pathogenic CNVs were identified in 29 patients (37.2%). Postnatal growth delay (p = 0.05564), pectus excavatum (p = 0.07484), brain imaging abnormalities (p = 0.07848), global developmental delay (p = 0.08070) and macrocephaly (p = 0.08919) were more likely to be associated with disease-causing CNVs.</p><p><strong>Conclusion: </strong>Our results allow phenotypic expansion of 14q11.2 microdeletions encompassing SUPT16H and CHD8 genes. Variants of unknown significance (n = 24) were found in 17 patients. We provide detailed phenotypic and genetic data of these individuals to facilitate future classification efforts, and spotlight two patients with potentially pathogenic alterations. Our results contribute to unraveling the diagnostic value of rare CNVs.</p>\",\"PeriodicalId\":19099,\"journal\":{\"name\":\"Molecular Cytogenetics\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":1.3000,\"publicationDate\":\"2022-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9623912/pdf/\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Cytogenetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13039-022-00623-z\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Cytogenetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13039-022-00623-z","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Clinical evaluation of rare copy number variations identified by chromosomal microarray in a Hungarian neurodevelopmental disorder patient cohort.
Background: Neurodevelopmental disorders are genetically heterogeneous pediatric conditions. The first tier diagnostic method for uncovering copy number variations (CNVs), one of the most common genetic etiologies in affected individuals, is chromosomal microarray (CMA). However, this methodology is not yet a routine molecular cytogenetic test in many parts of the world, including Hungary. Here we report clinical and genetic data of the first, relatively large Hungarian cohort of patients whose genetic testing included CMA.
Methods: Clinical data were retrospectively collected for 78 children who were analyzed using various CMA platforms. Phenotypes of patients with disease-causing variants were compared to patients with negative results using the chi squared/Fisher exact tests.
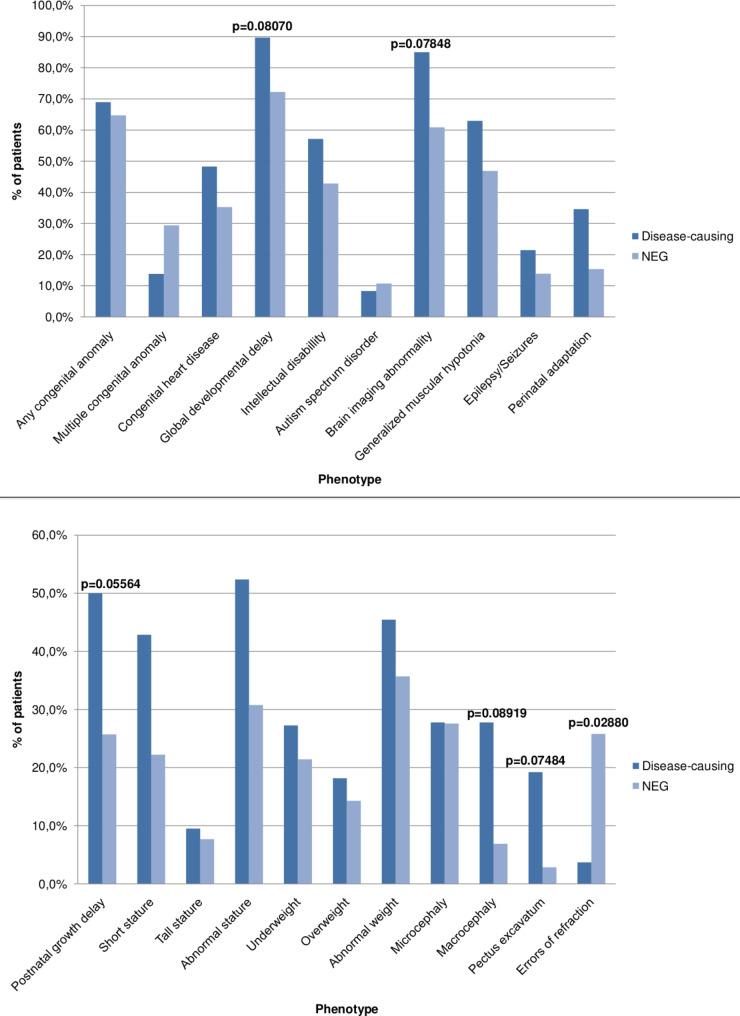
Results: A total of 30 pathogenic CNVs were identified in 29 patients (37.2%). Postnatal growth delay (p = 0.05564), pectus excavatum (p = 0.07484), brain imaging abnormalities (p = 0.07848), global developmental delay (p = 0.08070) and macrocephaly (p = 0.08919) were more likely to be associated with disease-causing CNVs.
Conclusion: Our results allow phenotypic expansion of 14q11.2 microdeletions encompassing SUPT16H and CHD8 genes. Variants of unknown significance (n = 24) were found in 17 patients. We provide detailed phenotypic and genetic data of these individuals to facilitate future classification efforts, and spotlight two patients with potentially pathogenic alterations. Our results contribute to unraveling the diagnostic value of rare CNVs.
期刊介绍:
Molecular Cytogenetics encompasses all aspects of chromosome biology and the application of molecular cytogenetic techniques in all areas of biology and medicine, including structural and functional organization of the chromosome and nucleus, genome variation, expression and evolution, chromosome abnormalities and genomic variations in medical genetics and tumor genetics.
Molecular Cytogenetics primarily defines a large set of the techniques that operate either with the entire genome or with specific targeted DNA sequences. Topical areas include, but are not limited to:
-Structural and functional organization of chromosome and nucleus-
Genome variation, expression and evolution-
Animal and plant molecular cytogenetics and genomics-
Chromosome abnormalities and genomic variations in clinical genetics-
Applications in preimplantation, pre- and post-natal diagnosis-
Applications in the central nervous system, cancer and haematology research-
Previously unreported applications of molecular cytogenetic techniques-
Development of new techniques or significant enhancements to established techniques.
This journal is a source for numerous scientists all over the world, who wish to improve or introduce molecular cytogenetic techniques into their practice.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
