Zhongyuan Zhao, Wei Liu, Gong Cheng, Shengjie Dong, Yuchi Zhao, Hao Wu, Zhilin Cao
{"title":"通过调节PEDF介导的NF-κB和NLRP3炎性体通路,敲除DAPK1可抑制IL-1β诱导的人软骨细胞炎症和软骨降解。","authors":"Zhongyuan Zhao, Wei Liu, Gong Cheng, Shengjie Dong, Yuchi Zhao, Hao Wu, Zhilin Cao","doi":"10.1177/17534259221086837","DOIUrl":null,"url":null,"abstract":"<p><p>Osteoarthritis (OA) is a common joint disease that is characterized by inflammation and cartilage degradation. Death-associated protein kinase 1 (DAPK1) is a multi-domain serine/threonine kinase and has been reported to be involved in the progression of OA. However, its role and mechanism in OA remain unclear. Here, we found the expression of DAPK1 in OA cartilage tissues was higher than that in normal cartilage tissues. The expression of DAPK1 in chondrocytes was up-regulated by IL-1β. Knockdown of DAPK1 promoted cell viability and anti-apoptotic protein expression, while it inhibited the apoptosis rate and pro-apoptotic protein expressions in IL-1β-induced chondrocytes. In addition, DAPK1 inhibition reduced the levels of inflammatory cytokines and expressions of matrix metalloproteinases (MMPs), and increased the expressions of collagen II and aggrecan. The data of mechanistic investigation indicated that the expression of pigment epithelium-derived factor (PEDF) was positively regulated by DAPK1. Overexpression of PEDF attenuated the effects of DAPK1 knockdown on IL-1β-induced cell viability, apoptosis, inflammation, and cartilage degradation. Furthermore, PEDF overexpression restored the activity of the NF-κB pathway and NLRP3 inflammasome after DAPK1 knockdown. Collectively, down-regulation of DAPK1 inhibited IL-1β-induced inflammation and cartilage degradation via the PEDF-mediated NF-κB and NLRP3 inflammasome pathways.</p>","PeriodicalId":13676,"journal":{"name":"Innate Immunity","volume":" ","pages":"21-30"},"PeriodicalIF":2.8000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10720599/pdf/","citationCount":"0","resultStr":"{\"title\":\"Knockdown of DAPK1 inhibits IL-1β-induced inflammation and cartilage degradation in human chondrocytes by modulating the PEDF-mediated NF-κB and NLRP3 inflammasome pathway.\",\"authors\":\"Zhongyuan Zhao, Wei Liu, Gong Cheng, Shengjie Dong, Yuchi Zhao, Hao Wu, Zhilin Cao\",\"doi\":\"10.1177/17534259221086837\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Osteoarthritis (OA) is a common joint disease that is characterized by inflammation and cartilage degradation. Death-associated protein kinase 1 (DAPK1) is a multi-domain serine/threonine kinase and has been reported to be involved in the progression of OA. However, its role and mechanism in OA remain unclear. Here, we found the expression of DAPK1 in OA cartilage tissues was higher than that in normal cartilage tissues. The expression of DAPK1 in chondrocytes was up-regulated by IL-1β. Knockdown of DAPK1 promoted cell viability and anti-apoptotic protein expression, while it inhibited the apoptosis rate and pro-apoptotic protein expressions in IL-1β-induced chondrocytes. In addition, DAPK1 inhibition reduced the levels of inflammatory cytokines and expressions of matrix metalloproteinases (MMPs), and increased the expressions of collagen II and aggrecan. The data of mechanistic investigation indicated that the expression of pigment epithelium-derived factor (PEDF) was positively regulated by DAPK1. Overexpression of PEDF attenuated the effects of DAPK1 knockdown on IL-1β-induced cell viability, apoptosis, inflammation, and cartilage degradation. Furthermore, PEDF overexpression restored the activity of the NF-κB pathway and NLRP3 inflammasome after DAPK1 knockdown. Collectively, down-regulation of DAPK1 inhibited IL-1β-induced inflammation and cartilage degradation via the PEDF-mediated NF-κB and NLRP3 inflammasome pathways.</p>\",\"PeriodicalId\":13676,\"journal\":{\"name\":\"Innate Immunity\",\"volume\":\" \",\"pages\":\"21-30\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2024-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10720599/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Innate Immunity\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1177/17534259221086837\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/11/21 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Innate Immunity","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1177/17534259221086837","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/11/21 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
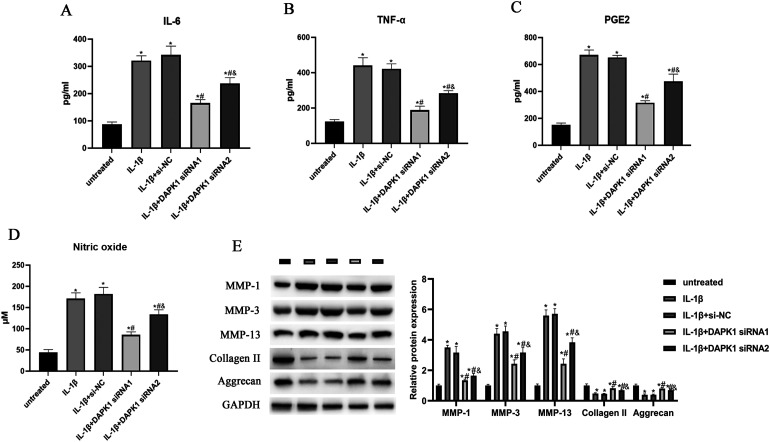
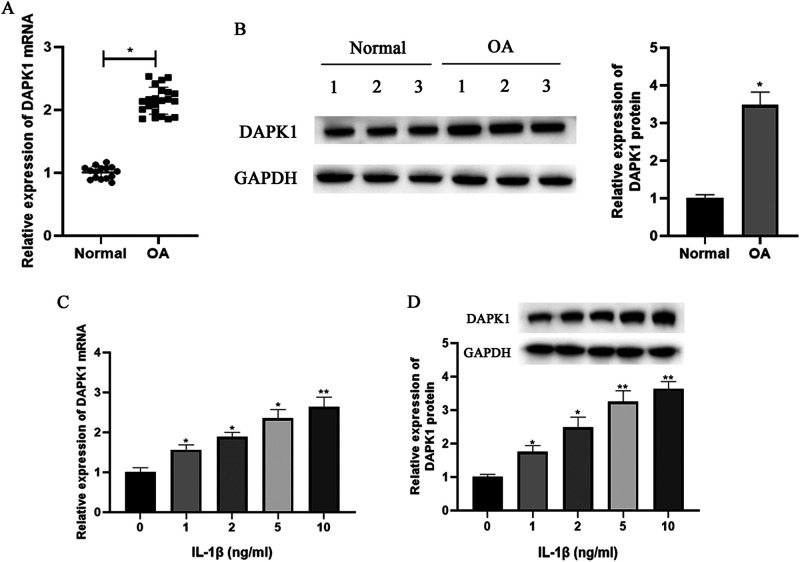
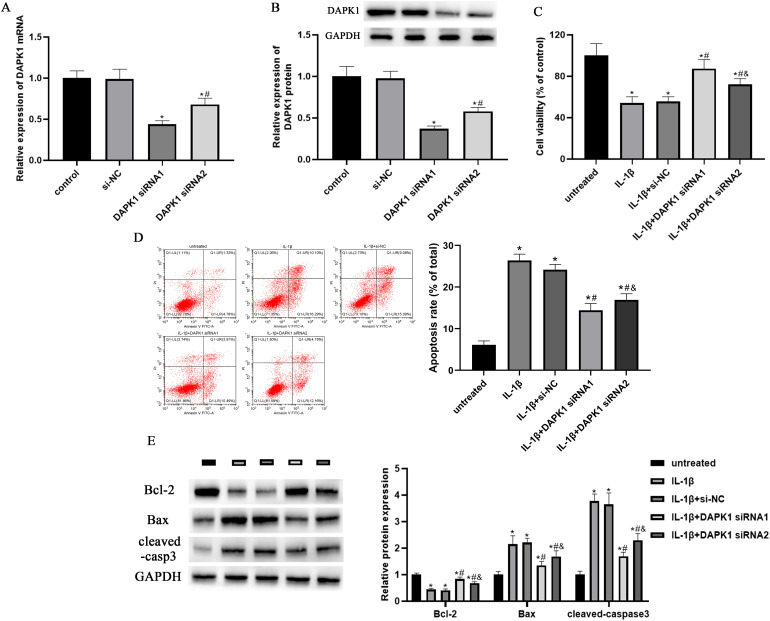
骨关节炎(OA)是一种以炎症和软骨退化为特征的常见关节疾病。死亡相关蛋白激酶1(DAPK1)是一种多域丝氨酸/苏氨酸激酶,有报道称它参与了OA的进展。然而,它在 OA 中的作用和机制仍不清楚。我们发现 DAPK1 在 OA 软骨组织中的表达高于正常软骨组织。DAPK1在软骨细胞中的表达受IL-1β上调。敲除DAPK1可促进细胞活力和抗凋亡蛋白的表达,同时抑制IL-1β诱导的软骨细胞的凋亡率和促凋亡蛋白的表达。此外,抑制 DAPK1 还能降低炎性细胞因子的水平和基质金属蛋白酶(MMPs)的表达,增加胶原蛋白 II 和 aggrecan 的表达。机理研究数据表明,色素上皮衍生因子(PEDF)的表达受 DAPK1 的正向调节。过量表达 PEDF 可减轻 DAPK1 敲除对 IL-1β 诱导的细胞活力、细胞凋亡、炎症和软骨降解的影响。此外,在 DAPK1 敲除后,PEDF 的过表达可恢复 NF-κB 通路和 NLRP3 炎性体的活性。总之,通过PEDF介导的NF-κB和NLRP3炎性体途径,下调DAPK1抑制了IL-1β诱导的炎症和软骨降解。
Knockdown of DAPK1 inhibits IL-1β-induced inflammation and cartilage degradation in human chondrocytes by modulating the PEDF-mediated NF-κB and NLRP3 inflammasome pathway.
Osteoarthritis (OA) is a common joint disease that is characterized by inflammation and cartilage degradation. Death-associated protein kinase 1 (DAPK1) is a multi-domain serine/threonine kinase and has been reported to be involved in the progression of OA. However, its role and mechanism in OA remain unclear. Here, we found the expression of DAPK1 in OA cartilage tissues was higher than that in normal cartilage tissues. The expression of DAPK1 in chondrocytes was up-regulated by IL-1β. Knockdown of DAPK1 promoted cell viability and anti-apoptotic protein expression, while it inhibited the apoptosis rate and pro-apoptotic protein expressions in IL-1β-induced chondrocytes. In addition, DAPK1 inhibition reduced the levels of inflammatory cytokines and expressions of matrix metalloproteinases (MMPs), and increased the expressions of collagen II and aggrecan. The data of mechanistic investigation indicated that the expression of pigment epithelium-derived factor (PEDF) was positively regulated by DAPK1. Overexpression of PEDF attenuated the effects of DAPK1 knockdown on IL-1β-induced cell viability, apoptosis, inflammation, and cartilage degradation. Furthermore, PEDF overexpression restored the activity of the NF-κB pathway and NLRP3 inflammasome after DAPK1 knockdown. Collectively, down-regulation of DAPK1 inhibited IL-1β-induced inflammation and cartilage degradation via the PEDF-mediated NF-κB and NLRP3 inflammasome pathways.
期刊介绍:
Innate Immunity is a highly ranked, peer-reviewed scholarly journal and is the official journal of the International Endotoxin & Innate Immunity Society (IEIIS). The journal welcomes manuscripts from researchers actively working on all aspects of innate immunity including biologically active bacterial, viral, fungal, parasitic, and plant components, as well as relevant cells, their receptors, signaling pathways, and induced mediators. The aim of the Journal is to provide a single, interdisciplinary forum for the dissemination of new information on innate immunity in humans, animals, and plants to researchers. The Journal creates a vehicle for the publication of articles encompassing all areas of research, basic, applied, and clinical. The subject areas of interest include, but are not limited to, research in biochemistry, biophysics, cell biology, chemistry, clinical medicine, immunology, infectious disease, microbiology, molecular biology, and pharmacology.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
