Prasanna Channathodiyil, Kieron May, Anne Segonds-Pichon, Paul D Smith, Simon J Cook, Jonathan Houseley
{"title":"在急性 MEK 抑制过程中摆脱 G1 停顿是获得耐药性的驱动因素。","authors":"Prasanna Channathodiyil, Kieron May, Anne Segonds-Pichon, Paul D Smith, Simon J Cook, Jonathan Houseley","doi":"10.1093/narcan/zcac032","DOIUrl":null,"url":null,"abstract":"<p><p>Mutations and gene amplifications that confer drug resistance emerge frequently during chemotherapy, but their mechanism and timing are poorly understood. Here, we investigate <i>BRAF<sup>V600E</sup></i> amplification events that underlie resistance to the MEK inhibitor selumetinib (AZD6244/ARRY-142886) in COLO205 cells, a well-characterized model for reproducible emergence of drug resistance, and show that <i>BRAF</i> amplifications acquired <i>de novo</i> are the primary cause of resistance. Selumetinib causes long-term G1 arrest accompanied by reduced expression of DNA replication and repair genes, but cells stochastically re-enter the cell cycle during treatment despite continued repression of pERK1/2. Most DNA replication and repair genes are re-expressed as cells enter S and G2; however, mRNAs encoding a subset of factors important for error-free replication and chromosome segregation, including TIPIN, PLK2 and PLK3, remain at low abundance. This suggests that DNA replication following escape from G1 arrest in drug is more error prone and provides a potential explanation for the DNA damage observed under long-term RAF-MEK-ERK1/2 pathway inhibition. To test the hypothesis that escape from G1 arrest in drug promotes <i>de novo BRAF</i> amplification, we exploited the combination of palbociclib and selumetinib. Combined treatment with selumetinib and a dose of palbociclib sufficient to reinforce G1 arrest in selumetinib-sensitive cells, but not to impair proliferation of resistant cells, delays the emergence of resistant colonies, meaning that escape from G1 arrest is critical in the formation of resistant clones. Our findings demonstrate that acquisition of MEK inhibitor resistance often occurs through <i>de novo</i> gene amplification and can be suppressed by impeding cell cycle entry in drug.</p>","PeriodicalId":18879,"journal":{"name":"NAR Cancer","volume":" ","pages":"zcac032"},"PeriodicalIF":0.0000,"publicationDate":"2022-10-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9575185/pdf/","citationCount":"0","resultStr":"{\"title\":\"Escape from G1 arrest during acute MEK inhibition drives the acquisition of drug resistance.\",\"authors\":\"Prasanna Channathodiyil, Kieron May, Anne Segonds-Pichon, Paul D Smith, Simon J Cook, Jonathan Houseley\",\"doi\":\"10.1093/narcan/zcac032\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Mutations and gene amplifications that confer drug resistance emerge frequently during chemotherapy, but their mechanism and timing are poorly understood. Here, we investigate <i>BRAF<sup>V600E</sup></i> amplification events that underlie resistance to the MEK inhibitor selumetinib (AZD6244/ARRY-142886) in COLO205 cells, a well-characterized model for reproducible emergence of drug resistance, and show that <i>BRAF</i> amplifications acquired <i>de novo</i> are the primary cause of resistance. Selumetinib causes long-term G1 arrest accompanied by reduced expression of DNA replication and repair genes, but cells stochastically re-enter the cell cycle during treatment despite continued repression of pERK1/2. Most DNA replication and repair genes are re-expressed as cells enter S and G2; however, mRNAs encoding a subset of factors important for error-free replication and chromosome segregation, including TIPIN, PLK2 and PLK3, remain at low abundance. This suggests that DNA replication following escape from G1 arrest in drug is more error prone and provides a potential explanation for the DNA damage observed under long-term RAF-MEK-ERK1/2 pathway inhibition. To test the hypothesis that escape from G1 arrest in drug promotes <i>de novo BRAF</i> amplification, we exploited the combination of palbociclib and selumetinib. Combined treatment with selumetinib and a dose of palbociclib sufficient to reinforce G1 arrest in selumetinib-sensitive cells, but not to impair proliferation of resistant cells, delays the emergence of resistant colonies, meaning that escape from G1 arrest is critical in the formation of resistant clones. Our findings demonstrate that acquisition of MEK inhibitor resistance often occurs through <i>de novo</i> gene amplification and can be suppressed by impeding cell cycle entry in drug.</p>\",\"PeriodicalId\":18879,\"journal\":{\"name\":\"NAR Cancer\",\"volume\":\" \",\"pages\":\"zcac032\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2022-10-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9575185/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NAR Cancer\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/narcan/zcac032\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/12/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NAR Cancer","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/narcan/zcac032","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/12/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
摘要
化疗过程中经常出现可产生耐药性的突变和基因扩增,但对其机制和时间却知之甚少。在这里,我们研究了 COLO205 细胞(一种具有良好特征的可重复出现耐药性的模型)中导致对 MEK 抑制剂塞卢米替(AZD6244/ARRY-142886)产生耐药性的 BRAFV600E 扩增事件,结果表明,从新获得的 BRAF 扩增是产生耐药性的主要原因。赛卢米替尼会导致细胞长期停滞在 G1 阶段,同时 DNA 复制和修复基因的表达也会减少,但尽管 pERK1/2 继续受到抑制,细胞仍会在治疗过程中随机地重新进入细胞周期。当细胞进入 S 期和 G2 期时,大多数 DNA 复制和修复基因都会重新表达;然而,编码对无差错复制和染色体分离非常重要的一组因子(包括 TIPIN、PLK2 和 PLK3)的 mRNA 仍然处于低丰度状态。这表明在药物中摆脱 G1 停顿后的 DNA 复制更容易出错,并为长期抑制 RAF-MEK-ERK1/2 通路时观察到的 DNA 损伤提供了潜在的解释。为了验证药物作用下摆脱 G1 停顿会促进新的 BRAF 扩增这一假设,我们利用了帕博西尼和赛鲁米替尼的联合治疗。塞卢米替尼和足以加强塞卢米替尼敏感细胞G1停滞、但不影响耐药细胞增殖的帕博西尼剂量联合治疗可延迟耐药菌落的出现,这意味着摆脱G1停滞对耐药克隆的形成至关重要。我们的研究结果表明,MEK抑制剂耐药性的获得往往是通过新基因扩增发生的,可以通过阻碍细胞周期进入药物来抑制。
Escape from G1 arrest during acute MEK inhibition drives the acquisition of drug resistance.
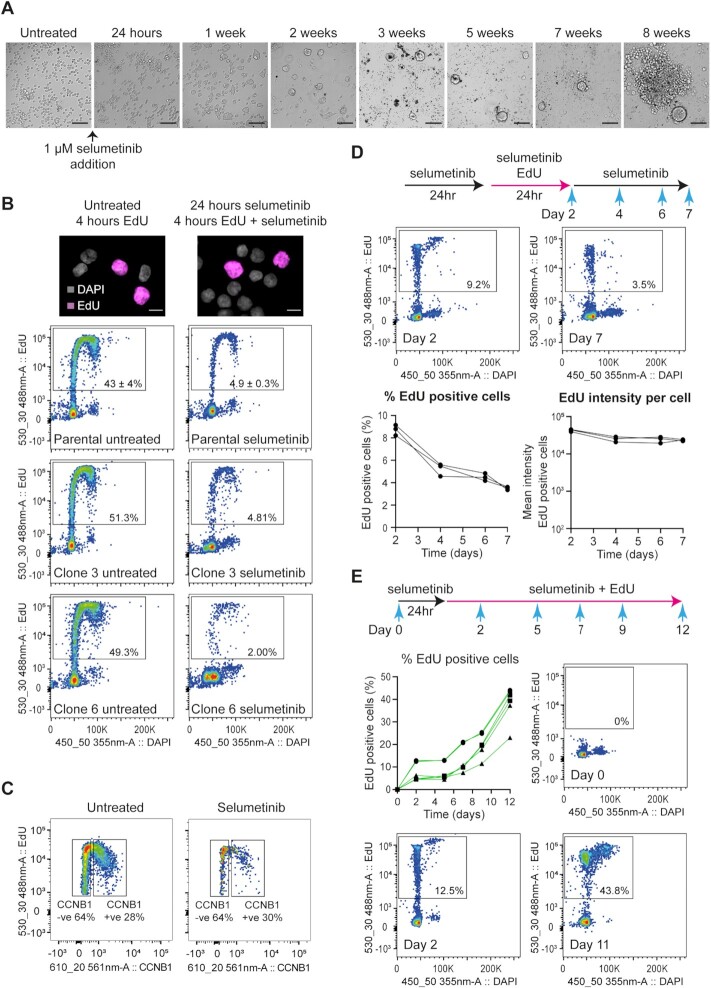
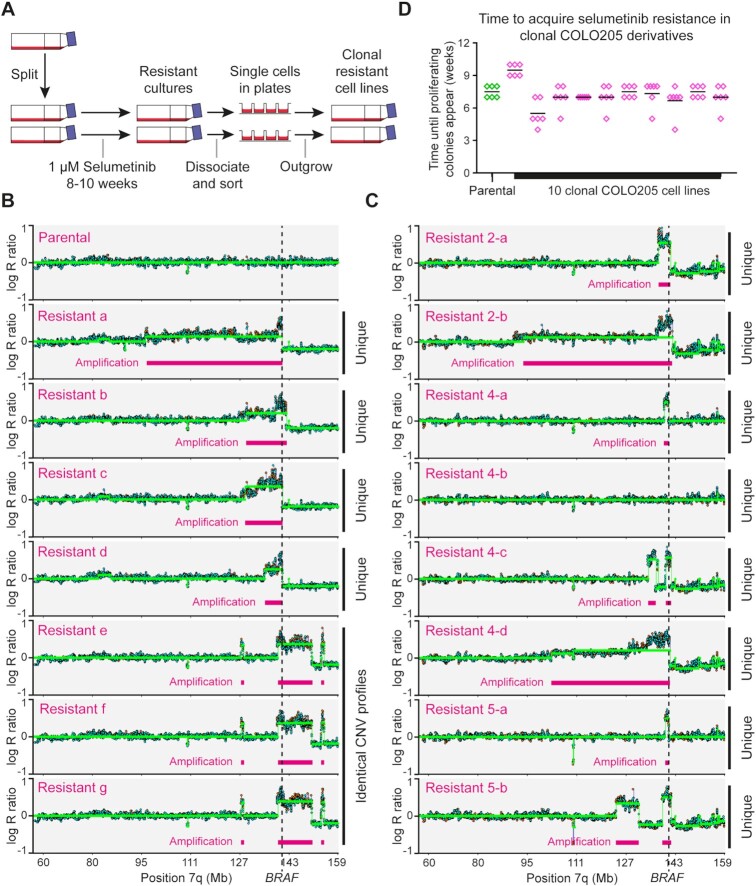
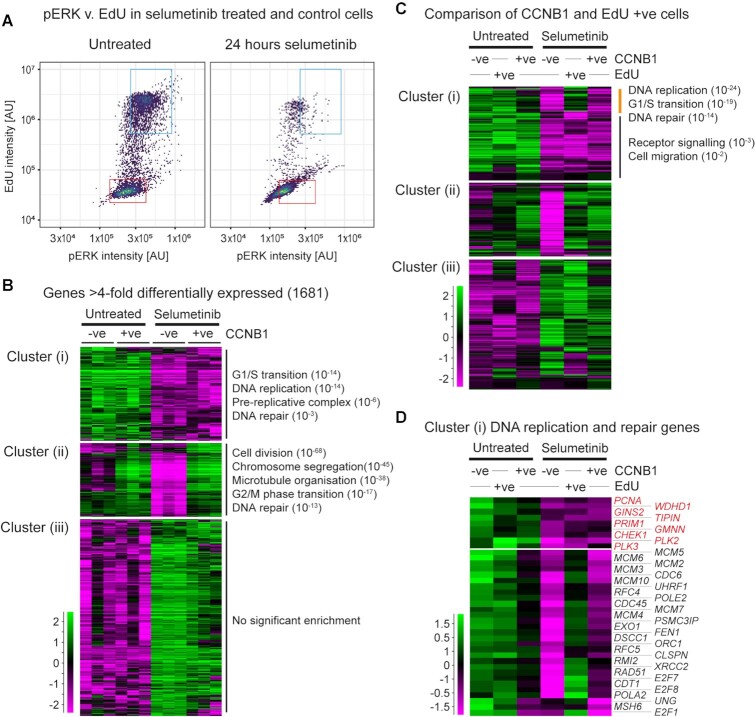
Mutations and gene amplifications that confer drug resistance emerge frequently during chemotherapy, but their mechanism and timing are poorly understood. Here, we investigate BRAFV600E amplification events that underlie resistance to the MEK inhibitor selumetinib (AZD6244/ARRY-142886) in COLO205 cells, a well-characterized model for reproducible emergence of drug resistance, and show that BRAF amplifications acquired de novo are the primary cause of resistance. Selumetinib causes long-term G1 arrest accompanied by reduced expression of DNA replication and repair genes, but cells stochastically re-enter the cell cycle during treatment despite continued repression of pERK1/2. Most DNA replication and repair genes are re-expressed as cells enter S and G2; however, mRNAs encoding a subset of factors important for error-free replication and chromosome segregation, including TIPIN, PLK2 and PLK3, remain at low abundance. This suggests that DNA replication following escape from G1 arrest in drug is more error prone and provides a potential explanation for the DNA damage observed under long-term RAF-MEK-ERK1/2 pathway inhibition. To test the hypothesis that escape from G1 arrest in drug promotes de novo BRAF amplification, we exploited the combination of palbociclib and selumetinib. Combined treatment with selumetinib and a dose of palbociclib sufficient to reinforce G1 arrest in selumetinib-sensitive cells, but not to impair proliferation of resistant cells, delays the emergence of resistant colonies, meaning that escape from G1 arrest is critical in the formation of resistant clones. Our findings demonstrate that acquisition of MEK inhibitor resistance often occurs through de novo gene amplification and can be suppressed by impeding cell cycle entry in drug.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
