Byung-Yong Park, Melanie Tachi-Duprat, Chibuike Ihewulezi, Arun Devotta, Jean-Pierre Saint-Jeannet
{"title":"核心剪接因子EFTUD2、SNRPB和TXNL4A对神经嵴和颅面发育至关重要。","authors":"Byung-Yong Park, Melanie Tachi-Duprat, Chibuike Ihewulezi, Arun Devotta, Jean-Pierre Saint-Jeannet","doi":"10.3390/jdb10030029","DOIUrl":null,"url":null,"abstract":"<p><p>Mandibulofacial dysostosis (MFD) is a human congenital disorder characterized by hypoplastic neural-crest-derived craniofacial bones often associated with outer and middle ear defects. There is growing evidence that mutations in components of the spliceosome are a major cause for MFD. Genetic variants affecting the function of several core splicing factors, namely <i>SF3B4</i>, <i>SF3B2</i>, <i>EFTUD2</i>, <i>SNRPB</i> and <i>TXNL4A</i>, are responsible for MFD in five related but distinct syndromes known as Nager and Rodriguez syndromes (NRS), craniofacial microsomia (CFM), mandibulofacial dysostosis with microcephaly (MFDM), cerebro-costo-mandibular syndrome (CCMS) and Burn-McKeown syndrome (BMKS), respectively. Animal models of NRS and MFDM indicate that MFD results from an early depletion of neural crest progenitors through a mechanism that involves apoptosis. Here we characterize the knockdown phenotype of Eftud2, Snrpb and Txnl4a in <i>Xenopus</i> embryos at different stages of neural crest and craniofacial development. Our results point to defects in cranial neural crest cell formation as the likely culprit for MFD associated with <i>EFTUD2</i>, <i>SNRPB</i> and <i>TXNL4A</i> haploinsufficiency, and suggest a commonality in the etiology of these craniofacial spliceosomopathies.</p>","PeriodicalId":15563,"journal":{"name":"Journal of Developmental Biology","volume":" ","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2022-07-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9326569/pdf/","citationCount":"3","resultStr":"{\"title\":\"The Core Splicing Factors EFTUD2, SNRPB and TXNL4A Are Essential for Neural Crest and Craniofacial Development.\",\"authors\":\"Byung-Yong Park, Melanie Tachi-Duprat, Chibuike Ihewulezi, Arun Devotta, Jean-Pierre Saint-Jeannet\",\"doi\":\"10.3390/jdb10030029\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Mandibulofacial dysostosis (MFD) is a human congenital disorder characterized by hypoplastic neural-crest-derived craniofacial bones often associated with outer and middle ear defects. There is growing evidence that mutations in components of the spliceosome are a major cause for MFD. Genetic variants affecting the function of several core splicing factors, namely <i>SF3B4</i>, <i>SF3B2</i>, <i>EFTUD2</i>, <i>SNRPB</i> and <i>TXNL4A</i>, are responsible for MFD in five related but distinct syndromes known as Nager and Rodriguez syndromes (NRS), craniofacial microsomia (CFM), mandibulofacial dysostosis with microcephaly (MFDM), cerebro-costo-mandibular syndrome (CCMS) and Burn-McKeown syndrome (BMKS), respectively. Animal models of NRS and MFDM indicate that MFD results from an early depletion of neural crest progenitors through a mechanism that involves apoptosis. Here we characterize the knockdown phenotype of Eftud2, Snrpb and Txnl4a in <i>Xenopus</i> embryos at different stages of neural crest and craniofacial development. Our results point to defects in cranial neural crest cell formation as the likely culprit for MFD associated with <i>EFTUD2</i>, <i>SNRPB</i> and <i>TXNL4A</i> haploinsufficiency, and suggest a commonality in the etiology of these craniofacial spliceosomopathies.</p>\",\"PeriodicalId\":15563,\"journal\":{\"name\":\"Journal of Developmental Biology\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2022-07-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9326569/pdf/\",\"citationCount\":\"3\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Developmental Biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3390/jdb10030029\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"DEVELOPMENTAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Developmental Biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/jdb10030029","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"DEVELOPMENTAL BIOLOGY","Score":null,"Total":0}
The Core Splicing Factors EFTUD2, SNRPB and TXNL4A Are Essential for Neural Crest and Craniofacial Development.
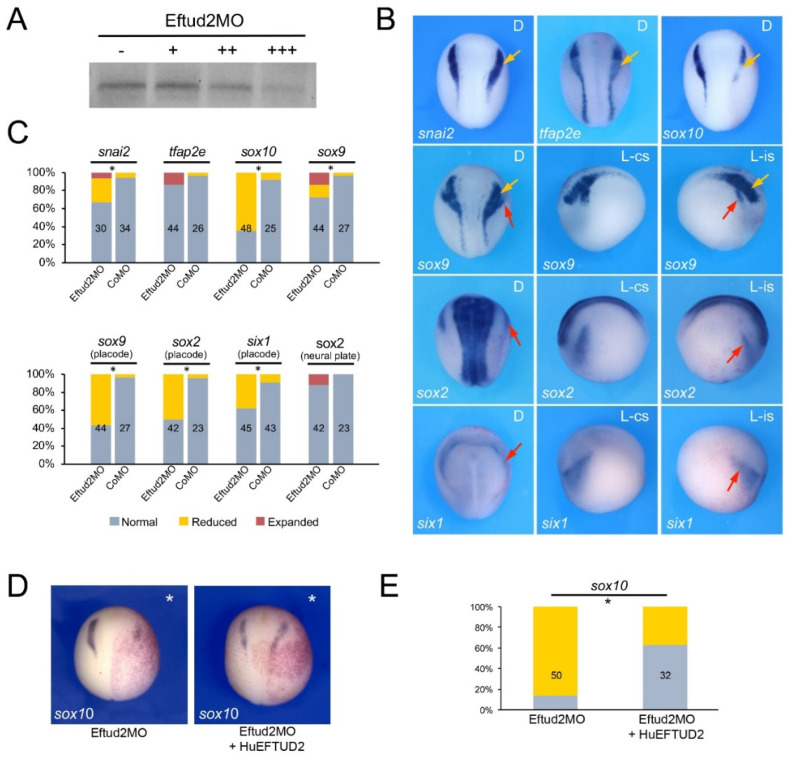
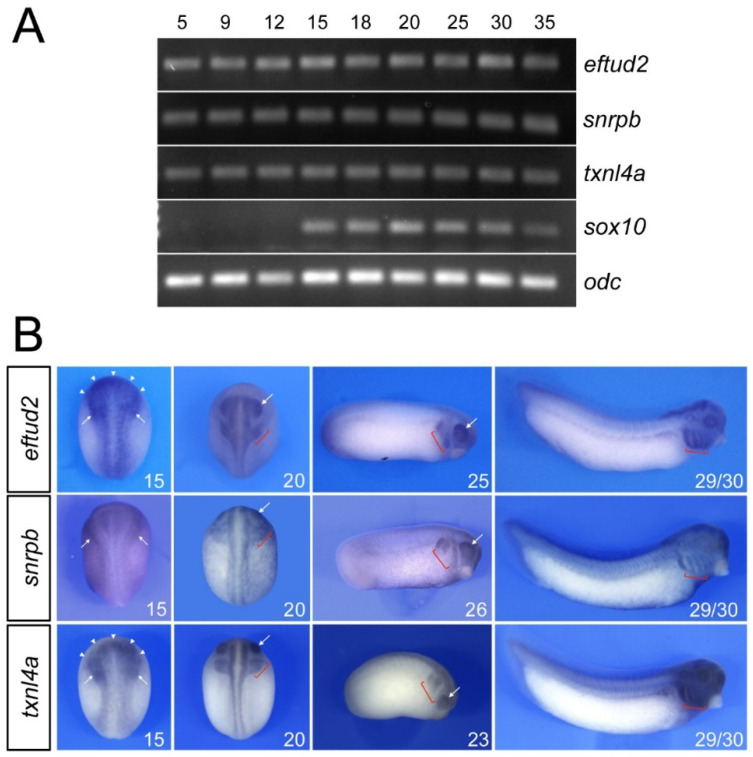
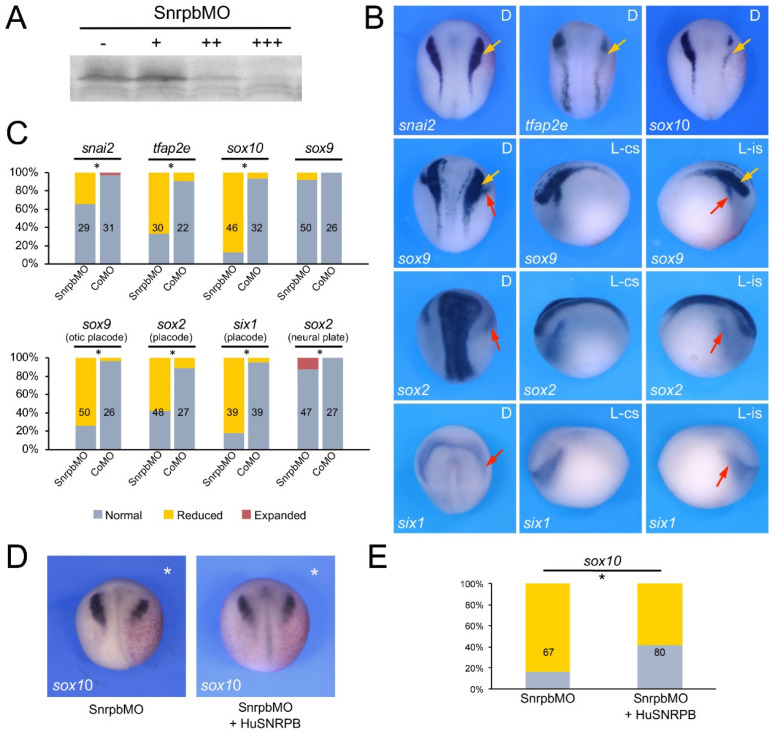
Mandibulofacial dysostosis (MFD) is a human congenital disorder characterized by hypoplastic neural-crest-derived craniofacial bones often associated with outer and middle ear defects. There is growing evidence that mutations in components of the spliceosome are a major cause for MFD. Genetic variants affecting the function of several core splicing factors, namely SF3B4, SF3B2, EFTUD2, SNRPB and TXNL4A, are responsible for MFD in five related but distinct syndromes known as Nager and Rodriguez syndromes (NRS), craniofacial microsomia (CFM), mandibulofacial dysostosis with microcephaly (MFDM), cerebro-costo-mandibular syndrome (CCMS) and Burn-McKeown syndrome (BMKS), respectively. Animal models of NRS and MFDM indicate that MFD results from an early depletion of neural crest progenitors through a mechanism that involves apoptosis. Here we characterize the knockdown phenotype of Eftud2, Snrpb and Txnl4a in Xenopus embryos at different stages of neural crest and craniofacial development. Our results point to defects in cranial neural crest cell formation as the likely culprit for MFD associated with EFTUD2, SNRPB and TXNL4A haploinsufficiency, and suggest a commonality in the etiology of these craniofacial spliceosomopathies.
期刊介绍:
The Journal of Developmental Biology (ISSN 2221-3759) is an international, peer-reviewed, quick-refereeing, open access journal, which publishes reviews, research papers and communications on the development of multicellular organisms at the molecule, cell, tissue, organ and whole organism levels. Our aim is to encourage researchers to effortlessly publish their new findings or concepts rapidly in an open access medium, overseen by their peers. There is no restriction on the length of the papers; the full experimental details must be provided so that the results can be reproduced. Electronic files regarding the full details of the experimental procedure, if unable to be published in a normal way, can be deposited as supplementary material. Journal of Developmental Biology focuses on: -Development mechanisms and genetics -Cell differentiation -Embryonal development -Tissue/organism growth -Metamorphosis and regeneration of the organisms. It involves many biological fields, such as Molecular biology, Genetics, Physiology, Cell biology, Anatomy, Embryology, Cancer research, Neurobiology, Immunology, Ecology, Evolutionary biology.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
