{"title":"与低髓鞘性白质营养不良2型疾病相关的GJC2新突变","authors":"Sajad Rafiee Komachali, Mozhgan Sheikholeslami, Mansoor Salehi","doi":"10.5808/gi.22008","DOIUrl":null,"url":null,"abstract":"<p><p>Hypomyelinating leukodystrophy type 2 (HLD2), is an inherited genetic disease of the central nervous system caused by recessive mutations in the gap junction protein gamma 2 (GJC2/GJA12). HLD2 is characterized by nystagmus, developmental delay, motor impairments, ataxia, severe speech problem, and hypomyelination in the brain. The GJC2 sequence encodes connexin 47 protein (Cx47). Connexins are a group of membrane proteins that oligomerize to construct gap junctions protein. In the present study, a novel missense mutation gene c.760G>A (p.Val254Met) was identified in a patient with HLD2 by performing whole exome sequencing. Following the discovery of the new mutation in the proband, we used Sanger sequencing to analyze his affected sibling and parents. Sanger sequencing verified homozygosity of the mutation in the proband and his affected sibling. The autosomal recessive inheritance pattern was confirmed since Sanger sequencing revealed both healthy parents were heterozygous for the mutation. PolyPhen2, SIFT, PROVEAN, and CADD were used to evaluate the function prediction scores of detected mutations. Cx47 is essential for oligodendrocyte function, including adequate myelination and myelin maintenance in humans. Novel mutation p.Val254Met is located in the second extracellular domain of Cx47, both extracellular loops are highly conserved and probably induce intramolecular disulfide interactions. This novel mutation in the Cx47 gene causes oligodendrocyte dysfunction and HLD2 disorder.</p>","PeriodicalId":36591,"journal":{"name":"Genomics and Informatics","volume":"20 2","pages":"e24"},"PeriodicalIF":0.0000,"publicationDate":"2022-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9299563/pdf/","citationCount":"1","resultStr":"{\"title\":\"A novel mutation in GJC2 associated with hypomyelinating leukodystrophy type 2 disorder.\",\"authors\":\"Sajad Rafiee Komachali, Mozhgan Sheikholeslami, Mansoor Salehi\",\"doi\":\"10.5808/gi.22008\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Hypomyelinating leukodystrophy type 2 (HLD2), is an inherited genetic disease of the central nervous system caused by recessive mutations in the gap junction protein gamma 2 (GJC2/GJA12). HLD2 is characterized by nystagmus, developmental delay, motor impairments, ataxia, severe speech problem, and hypomyelination in the brain. The GJC2 sequence encodes connexin 47 protein (Cx47). Connexins are a group of membrane proteins that oligomerize to construct gap junctions protein. In the present study, a novel missense mutation gene c.760G>A (p.Val254Met) was identified in a patient with HLD2 by performing whole exome sequencing. Following the discovery of the new mutation in the proband, we used Sanger sequencing to analyze his affected sibling and parents. Sanger sequencing verified homozygosity of the mutation in the proband and his affected sibling. The autosomal recessive inheritance pattern was confirmed since Sanger sequencing revealed both healthy parents were heterozygous for the mutation. PolyPhen2, SIFT, PROVEAN, and CADD were used to evaluate the function prediction scores of detected mutations. Cx47 is essential for oligodendrocyte function, including adequate myelination and myelin maintenance in humans. Novel mutation p.Val254Met is located in the second extracellular domain of Cx47, both extracellular loops are highly conserved and probably induce intramolecular disulfide interactions. This novel mutation in the Cx47 gene causes oligodendrocyte dysfunction and HLD2 disorder.</p>\",\"PeriodicalId\":36591,\"journal\":{\"name\":\"Genomics and Informatics\",\"volume\":\"20 2\",\"pages\":\"e24\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2022-06-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9299563/pdf/\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genomics and Informatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.5808/gi.22008\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/6/30 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"Agricultural and Biological Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genomics and Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5808/gi.22008","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/6/30 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
引用次数: 1
摘要
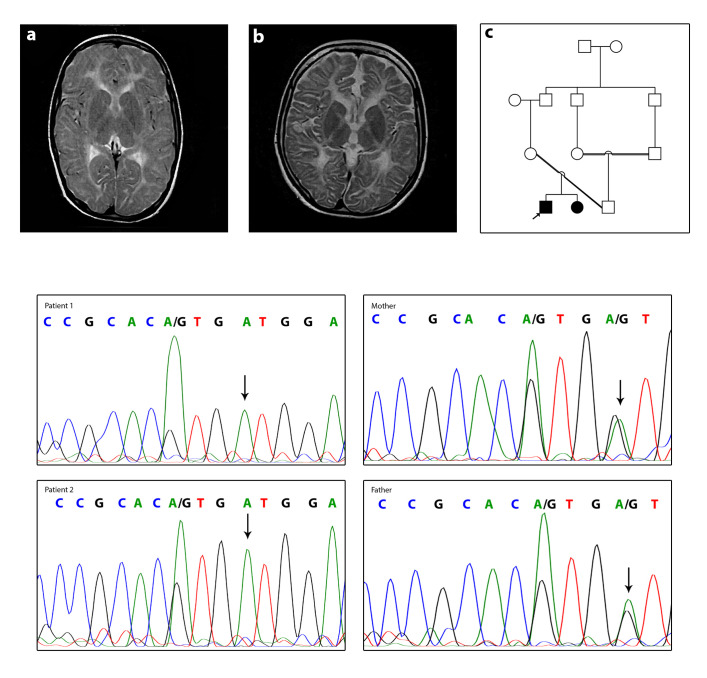
2型低髓鞘性白质营养不良症(HLD2)是一种由间隙连接蛋白γ 2 (GJC2/GJA12)隐性突变引起的中枢神经系统遗传性疾病。HLD2的特点是眼球震颤、发育迟缓、运动障碍、共济失调、严重的语言问题和大脑髓鞘发育低下。GJC2序列编码连接蛋白47 (Cx47)。连接蛋白是一组寡聚形成间隙连接蛋白的膜蛋白。在本研究中,通过对HLD2患者进行全外显子组测序,发现了一种新的错义突变基因c.760G> a (p.Val254Met)。在先证者身上发现新的突变后,我们用桑格测序法分析了他受影响的兄弟姐妹和父母。桑格测序证实了先证者及其患病兄弟姐妹突变的纯合性。常染色体隐性遗传模式被证实,因为Sanger测序显示健康的父母都是杂合突变。使用PolyPhen2、SIFT、PROVEAN和CADD评估检测到的突变的功能预测评分。Cx47对人类少突胶质细胞功能至关重要,包括充分的髓鞘形成和髓磷脂维持。新突变p.Val254Met位于Cx47的第二个胞外结构域,两个胞外环高度保守,可能诱导分子内二硫相互作用。Cx47基因的这种新突变导致少突胶质细胞功能障碍和HLD2疾病。
A novel mutation in GJC2 associated with hypomyelinating leukodystrophy type 2 disorder.
Hypomyelinating leukodystrophy type 2 (HLD2), is an inherited genetic disease of the central nervous system caused by recessive mutations in the gap junction protein gamma 2 (GJC2/GJA12). HLD2 is characterized by nystagmus, developmental delay, motor impairments, ataxia, severe speech problem, and hypomyelination in the brain. The GJC2 sequence encodes connexin 47 protein (Cx47). Connexins are a group of membrane proteins that oligomerize to construct gap junctions protein. In the present study, a novel missense mutation gene c.760G>A (p.Val254Met) was identified in a patient with HLD2 by performing whole exome sequencing. Following the discovery of the new mutation in the proband, we used Sanger sequencing to analyze his affected sibling and parents. Sanger sequencing verified homozygosity of the mutation in the proband and his affected sibling. The autosomal recessive inheritance pattern was confirmed since Sanger sequencing revealed both healthy parents were heterozygous for the mutation. PolyPhen2, SIFT, PROVEAN, and CADD were used to evaluate the function prediction scores of detected mutations. Cx47 is essential for oligodendrocyte function, including adequate myelination and myelin maintenance in humans. Novel mutation p.Val254Met is located in the second extracellular domain of Cx47, both extracellular loops are highly conserved and probably induce intramolecular disulfide interactions. This novel mutation in the Cx47 gene causes oligodendrocyte dysfunction and HLD2 disorder.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
