{"title":"HPRT1基因临床表型及功能的研究。","authors":"Miao Guo, Yucai Chen, Longlong Lin, Yilin Wang, Anqi Wang, Fang Yuan, Chunmei Wang, Simei Wang, Yuanfeng Zhang","doi":"10.1177/2329048X221108821","DOIUrl":null,"url":null,"abstract":"<p><p><b>Background:</b> Lesch-Nyhan disease (LND) is a rare x-linked purine metabolic neurogenetic disease caused by enzyme hypoxanthine-guanine phosphoriribosyltransferase(HGprt) deficiency, also known as self-destructive appearance syndrome. A series of manifestations are caused by abnormal purine metabolism. The typical clinical manifestations are hyperuricemia, growth retardation, mental retardation, short stature, dance-like athetosis, aggressive behavior, and compulsive self-harm. <b>Methods:</b> We identified a point mutation c.151C > T (p. Arg51*) in a pedigree. We analyzed the clinical characteristics of children in a family, and obtained the blood of their parents and siblings for second-generation sequencing. At the same time, we also analyzed and compared the expression of HPRT1 gene and predicted the three-dimensional structure of the protein. And we analyzed the clinical manifestations caused by the defect of the HPRT1 gene. <b>Results:</b> The mutation led to the termination of transcription at the 51st arginine, resulting in the production of truncated protein, and the relative expression of HPRT1 gene in patients was significantly lower than other family members and 10 normal individuals. <b>Conclusion:</b> This mutation leads to the early termination of protein translation and the formation of a truncated HPRT protein, which affects the function of the protein and generates corresponding clinical manifestations.</p>","PeriodicalId":72572,"journal":{"name":"Child neurology open","volume":" ","pages":"2329048X221108821"},"PeriodicalIF":0.0000,"publicationDate":"2022-07-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/1c/07/10.1177_2329048X221108821.PMC9305801.pdf","citationCount":"1","resultStr":"{\"title\":\"The Study on the Clinical Phenotype and Function of HPRT1 Gene.\",\"authors\":\"Miao Guo, Yucai Chen, Longlong Lin, Yilin Wang, Anqi Wang, Fang Yuan, Chunmei Wang, Simei Wang, Yuanfeng Zhang\",\"doi\":\"10.1177/2329048X221108821\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p><b>Background:</b> Lesch-Nyhan disease (LND) is a rare x-linked purine metabolic neurogenetic disease caused by enzyme hypoxanthine-guanine phosphoriribosyltransferase(HGprt) deficiency, also known as self-destructive appearance syndrome. A series of manifestations are caused by abnormal purine metabolism. The typical clinical manifestations are hyperuricemia, growth retardation, mental retardation, short stature, dance-like athetosis, aggressive behavior, and compulsive self-harm. <b>Methods:</b> We identified a point mutation c.151C > T (p. Arg51*) in a pedigree. We analyzed the clinical characteristics of children in a family, and obtained the blood of their parents and siblings for second-generation sequencing. At the same time, we also analyzed and compared the expression of HPRT1 gene and predicted the three-dimensional structure of the protein. And we analyzed the clinical manifestations caused by the defect of the HPRT1 gene. <b>Results:</b> The mutation led to the termination of transcription at the 51st arginine, resulting in the production of truncated protein, and the relative expression of HPRT1 gene in patients was significantly lower than other family members and 10 normal individuals. <b>Conclusion:</b> This mutation leads to the early termination of protein translation and the formation of a truncated HPRT protein, which affects the function of the protein and generates corresponding clinical manifestations.</p>\",\"PeriodicalId\":72572,\"journal\":{\"name\":\"Child neurology open\",\"volume\":\" \",\"pages\":\"2329048X221108821\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2022-07-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/1c/07/10.1177_2329048X221108821.PMC9305801.pdf\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Child neurology open\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/2329048X221108821\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Child neurology open","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/2329048X221108821","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 1
摘要
背景:Lesch-Nyhan病(LND)是一种罕见的x连锁嘌呤代谢性神经遗传疾病,由次黄嘌呤-鸟嘌呤磷酸核糖基转移酶(HGprt)缺乏引起,也称为自毁外观综合征。嘌呤代谢异常引起的一系列表现。典型临床表现为高尿酸血症、生长发育迟缓、智力低下、身材矮小、舞蹈样手足动症、攻击性行为、强迫性自残。方法:在一个家系中发现一个点突变c.151C > T (p. Arg51*)。我们分析了一个家庭中儿童的临床特征,并获得了其父母和兄弟姐妹的血液进行二代测序。同时,我们还分析比较了HPRT1基因的表达情况,并预测了该蛋白的三维结构。并分析了HPRT1基因缺陷引起的临床表现。结果:突变导致51号精氨酸转录终止,产生截断蛋白,患者中HPRT1基因的相对表达量显著低于其他家族成员和10名正常人。结论:该突变导致蛋白翻译提前终止,形成截断的HPRT蛋白,影响蛋白功能,产生相应的临床表现。
The Study on the Clinical Phenotype and Function of HPRT1 Gene.
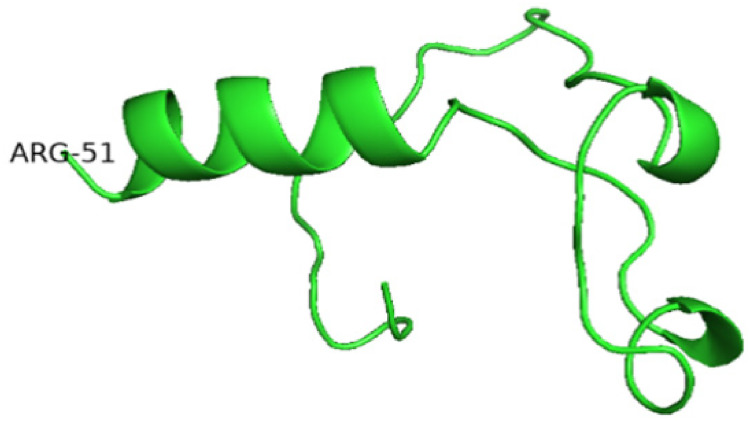
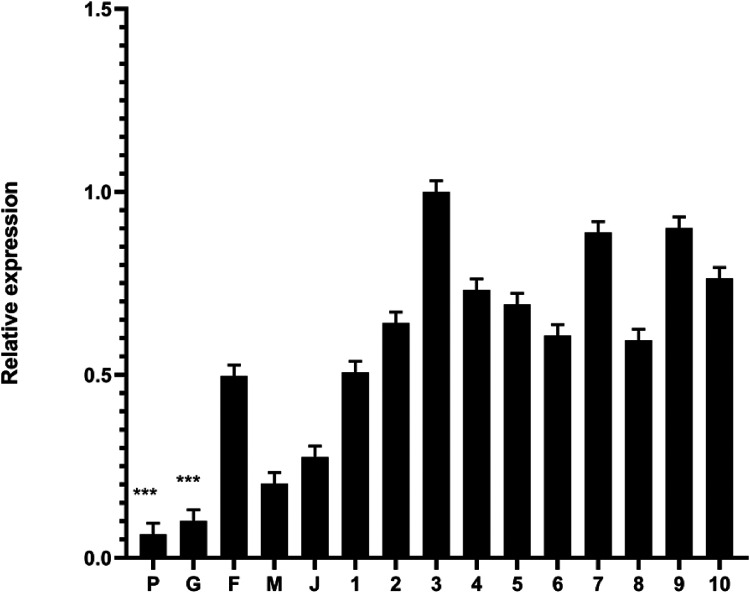
Background: Lesch-Nyhan disease (LND) is a rare x-linked purine metabolic neurogenetic disease caused by enzyme hypoxanthine-guanine phosphoriribosyltransferase(HGprt) deficiency, also known as self-destructive appearance syndrome. A series of manifestations are caused by abnormal purine metabolism. The typical clinical manifestations are hyperuricemia, growth retardation, mental retardation, short stature, dance-like athetosis, aggressive behavior, and compulsive self-harm. Methods: We identified a point mutation c.151C > T (p. Arg51*) in a pedigree. We analyzed the clinical characteristics of children in a family, and obtained the blood of their parents and siblings for second-generation sequencing. At the same time, we also analyzed and compared the expression of HPRT1 gene and predicted the three-dimensional structure of the protein. And we analyzed the clinical manifestations caused by the defect of the HPRT1 gene. Results: The mutation led to the termination of transcription at the 51st arginine, resulting in the production of truncated protein, and the relative expression of HPRT1 gene in patients was significantly lower than other family members and 10 normal individuals. Conclusion: This mutation leads to the early termination of protein translation and the formation of a truncated HPRT protein, which affects the function of the protein and generates corresponding clinical manifestations.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
