Davide Chicco, Abbas Alameer, Sara Rahmati, Giuseppe Jurman
{"title":"基于问题集和集成机器学习的基因表达的潜在泛癌症预后标记。","authors":"Davide Chicco, Abbas Alameer, Sara Rahmati, Giuseppe Jurman","doi":"10.1186/s13040-022-00312-y","DOIUrl":null,"url":null,"abstract":"<p><p>Cancer is one of the leading causes of death worldwide and can be caused by environmental aspects (for example, exposure to asbestos), by human behavior (such as smoking), or by genetic factors. To understand which genes might be involved in patients' survival, researchers have invented prognostic genetic signatures: lists of genes that can be used in scientific analyses to predict if a patient will survive or not. In this study, we joined together five different prognostic signatures, each of them related to a specific cancer type, to generate a unique pan-cancer prognostic signature, that contains 207 unique probesets related to 187 unique gene symbols, with one particular probeset present in two cancer type-specific signatures (203072_at related to the MYO1E gene). We applied our proposed pan-cancer signature with the Random Forests machine learning method to 57 microarray gene expression datasets of 12 different cancer types, and analyzed the results. We also compared the performance of our pan-cancer signature with the performances of two alternative prognostic signatures, and with the performances of each cancer type-specific signature on their corresponding cancer type-specific datasets. Our results confirmed the effectiveness of our prognostic pan-cancer signature. Moreover, we performed a pathway enrichment analysis, which indicated an association between the signature genes and a protein-protein interaction analysis, that highlighted PIK3R2 and FN1 as key genes having a fundamental relevance in our signature, suggesting an important role in pan-cancer prognosis for both of them.</p>","PeriodicalId":48947,"journal":{"name":"Biodata Mining","volume":" ","pages":"28"},"PeriodicalIF":6.1000,"publicationDate":"2022-11-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9632055/pdf/","citationCount":"1","resultStr":"{\"title\":\"Towards a potential pan-cancer prognostic signature for gene expression based on probesets and ensemble machine learning.\",\"authors\":\"Davide Chicco, Abbas Alameer, Sara Rahmati, Giuseppe Jurman\",\"doi\":\"10.1186/s13040-022-00312-y\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Cancer is one of the leading causes of death worldwide and can be caused by environmental aspects (for example, exposure to asbestos), by human behavior (such as smoking), or by genetic factors. To understand which genes might be involved in patients' survival, researchers have invented prognostic genetic signatures: lists of genes that can be used in scientific analyses to predict if a patient will survive or not. In this study, we joined together five different prognostic signatures, each of them related to a specific cancer type, to generate a unique pan-cancer prognostic signature, that contains 207 unique probesets related to 187 unique gene symbols, with one particular probeset present in two cancer type-specific signatures (203072_at related to the MYO1E gene). We applied our proposed pan-cancer signature with the Random Forests machine learning method to 57 microarray gene expression datasets of 12 different cancer types, and analyzed the results. We also compared the performance of our pan-cancer signature with the performances of two alternative prognostic signatures, and with the performances of each cancer type-specific signature on their corresponding cancer type-specific datasets. Our results confirmed the effectiveness of our prognostic pan-cancer signature. Moreover, we performed a pathway enrichment analysis, which indicated an association between the signature genes and a protein-protein interaction analysis, that highlighted PIK3R2 and FN1 as key genes having a fundamental relevance in our signature, suggesting an important role in pan-cancer prognosis for both of them.</p>\",\"PeriodicalId\":48947,\"journal\":{\"name\":\"Biodata Mining\",\"volume\":\" \",\"pages\":\"28\"},\"PeriodicalIF\":6.1000,\"publicationDate\":\"2022-11-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9632055/pdf/\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biodata Mining\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13040-022-00312-y\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biodata Mining","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13040-022-00312-y","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Towards a potential pan-cancer prognostic signature for gene expression based on probesets and ensemble machine learning.
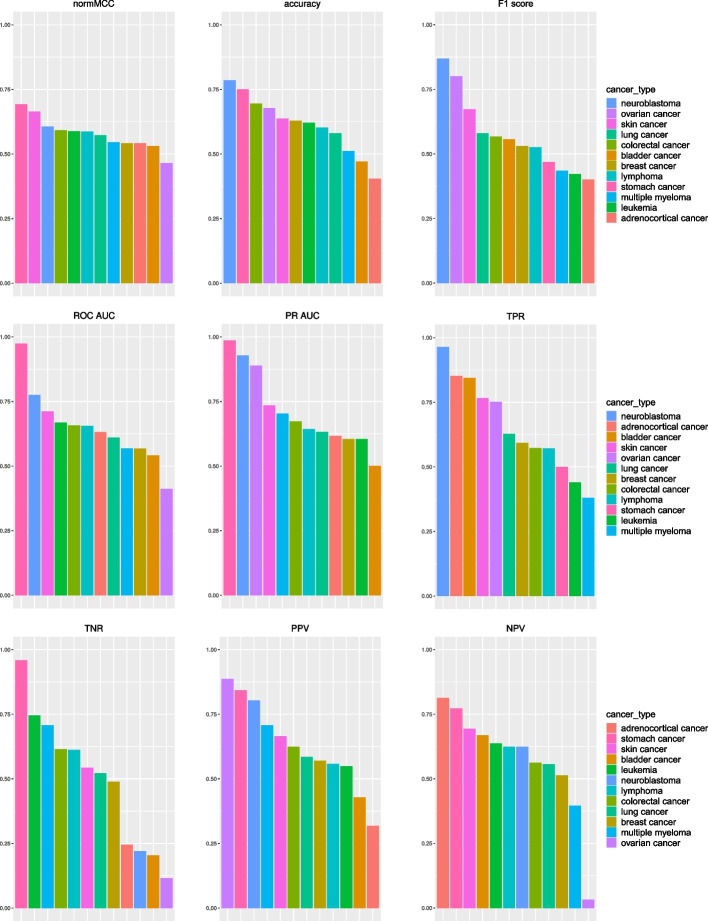
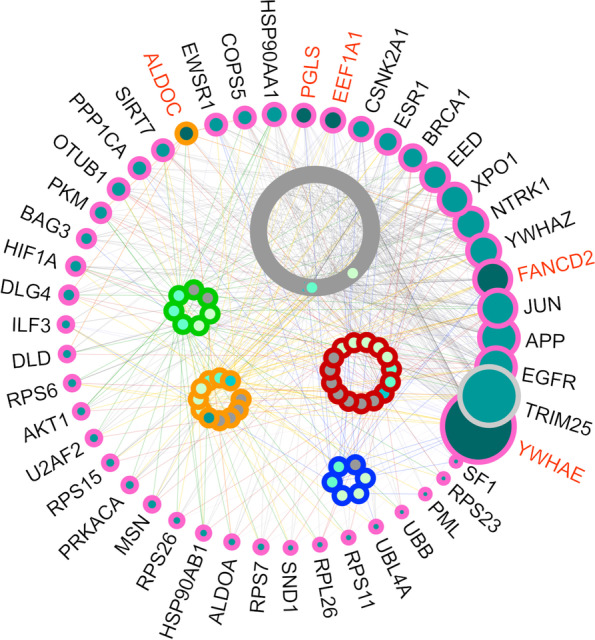
Cancer is one of the leading causes of death worldwide and can be caused by environmental aspects (for example, exposure to asbestos), by human behavior (such as smoking), or by genetic factors. To understand which genes might be involved in patients' survival, researchers have invented prognostic genetic signatures: lists of genes that can be used in scientific analyses to predict if a patient will survive or not. In this study, we joined together five different prognostic signatures, each of them related to a specific cancer type, to generate a unique pan-cancer prognostic signature, that contains 207 unique probesets related to 187 unique gene symbols, with one particular probeset present in two cancer type-specific signatures (203072_at related to the MYO1E gene). We applied our proposed pan-cancer signature with the Random Forests machine learning method to 57 microarray gene expression datasets of 12 different cancer types, and analyzed the results. We also compared the performance of our pan-cancer signature with the performances of two alternative prognostic signatures, and with the performances of each cancer type-specific signature on their corresponding cancer type-specific datasets. Our results confirmed the effectiveness of our prognostic pan-cancer signature. Moreover, we performed a pathway enrichment analysis, which indicated an association between the signature genes and a protein-protein interaction analysis, that highlighted PIK3R2 and FN1 as key genes having a fundamental relevance in our signature, suggesting an important role in pan-cancer prognosis for both of them.
期刊介绍:
BioData Mining is an open access, open peer-reviewed journal encompassing research on all aspects of data mining applied to high-dimensional biological and biomedical data, focusing on computational aspects of knowledge discovery from large-scale genetic, transcriptomic, genomic, proteomic, and metabolomic data.
Topical areas include, but are not limited to:
-Development, evaluation, and application of novel data mining and machine learning algorithms.
-Adaptation, evaluation, and application of traditional data mining and machine learning algorithms.
-Open-source software for the application of data mining and machine learning algorithms.
-Design, development and integration of databases, software and web services for the storage, management, retrieval, and analysis of data from large scale studies.
-Pre-processing, post-processing, modeling, and interpretation of data mining and machine learning results for biological interpretation and knowledge discovery.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
