{"title":"中枢性性早熟的遗传原因。","authors":"Toshihiro Tajima","doi":"10.1297/cpe.2022-0021","DOIUrl":null,"url":null,"abstract":"<p><p>Central precocious puberty (CPP) is a condition in which the hypothalamus-pituitary-gonadal system is activated earlier than the normal developmental stage. The etiology includes organic lesions in the brain; however, in the case of idiopathic diseases, environmental and/or genetic factors are involved in the development of CPP. A genetic abnormality in <i>KISS1R</i>, that encodes the kisspeptin receptor, was first reported in 2008 as a cause of idiopathic CPP. Furthermore, genetic alterations in <i>KISS1</i>, <i>MKRN3</i>, <i>DLK1</i>, and <i>PROKR2</i> have been reported in idiopathic and/or familial CPP. Of these, <i>MKRN3</i> has the highest frequency of pathological variants associated with CPP worldwide; but, abnormalities in <i>MKRN3</i> are rare in patients in East Asia, including Japan. <i>MKRN3</i> and <i>DLK1</i> are maternal imprinting genes; thus, CPP develops when a pathological variant is inherited from the father. The mechanism of CPP due to defects in <i>MKRN3</i> and <i>DLK1</i> has not been completely clarified, but it is suggested that both may negatively control the progression of puberty. CPP due to such a single gene abnormality is extremely rare, but it is important to understand the mechanisms of puberty and reproduction. A further development in the genetics of CPP is expected in the future.</p>","PeriodicalId":10678,"journal":{"name":"Clinical Pediatric Endocrinology","volume":"31 3","pages":"101-109"},"PeriodicalIF":1.2000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/74/4c/cpe-31-101.PMC9297165.pdf","citationCount":"4","resultStr":"{\"title\":\"Genetic causes of central precocious puberty.\",\"authors\":\"Toshihiro Tajima\",\"doi\":\"10.1297/cpe.2022-0021\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Central precocious puberty (CPP) is a condition in which the hypothalamus-pituitary-gonadal system is activated earlier than the normal developmental stage. The etiology includes organic lesions in the brain; however, in the case of idiopathic diseases, environmental and/or genetic factors are involved in the development of CPP. A genetic abnormality in <i>KISS1R</i>, that encodes the kisspeptin receptor, was first reported in 2008 as a cause of idiopathic CPP. Furthermore, genetic alterations in <i>KISS1</i>, <i>MKRN3</i>, <i>DLK1</i>, and <i>PROKR2</i> have been reported in idiopathic and/or familial CPP. Of these, <i>MKRN3</i> has the highest frequency of pathological variants associated with CPP worldwide; but, abnormalities in <i>MKRN3</i> are rare in patients in East Asia, including Japan. <i>MKRN3</i> and <i>DLK1</i> are maternal imprinting genes; thus, CPP develops when a pathological variant is inherited from the father. The mechanism of CPP due to defects in <i>MKRN3</i> and <i>DLK1</i> has not been completely clarified, but it is suggested that both may negatively control the progression of puberty. CPP due to such a single gene abnormality is extremely rare, but it is important to understand the mechanisms of puberty and reproduction. A further development in the genetics of CPP is expected in the future.</p>\",\"PeriodicalId\":10678,\"journal\":{\"name\":\"Clinical Pediatric Endocrinology\",\"volume\":\"31 3\",\"pages\":\"101-109\"},\"PeriodicalIF\":1.2000,\"publicationDate\":\"2022-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/74/4c/cpe-31-101.PMC9297165.pdf\",\"citationCount\":\"4\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Pediatric Endocrinology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1297/cpe.2022-0021\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/5/29 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Pediatric Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1297/cpe.2022-0021","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/5/29 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
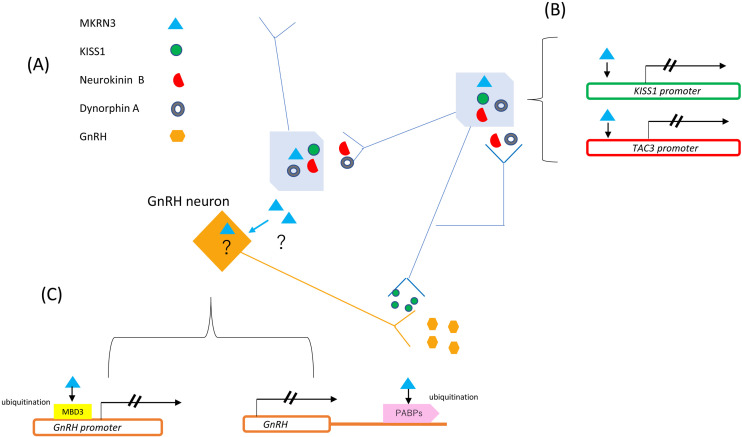
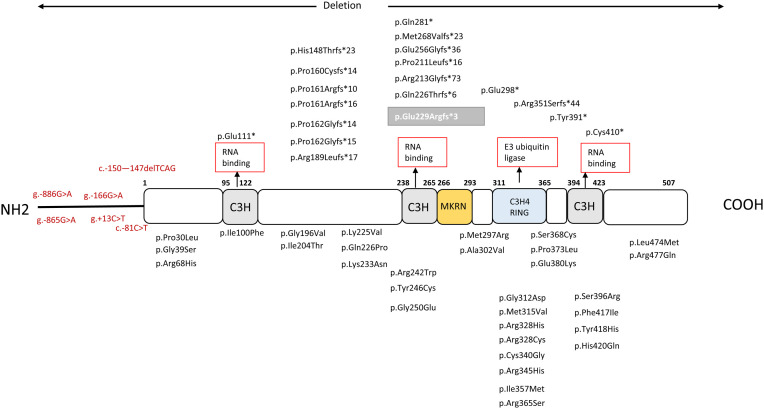
Central precocious puberty (CPP) is a condition in which the hypothalamus-pituitary-gonadal system is activated earlier than the normal developmental stage. The etiology includes organic lesions in the brain; however, in the case of idiopathic diseases, environmental and/or genetic factors are involved in the development of CPP. A genetic abnormality in KISS1R, that encodes the kisspeptin receptor, was first reported in 2008 as a cause of idiopathic CPP. Furthermore, genetic alterations in KISS1, MKRN3, DLK1, and PROKR2 have been reported in idiopathic and/or familial CPP. Of these, MKRN3 has the highest frequency of pathological variants associated with CPP worldwide; but, abnormalities in MKRN3 are rare in patients in East Asia, including Japan. MKRN3 and DLK1 are maternal imprinting genes; thus, CPP develops when a pathological variant is inherited from the father. The mechanism of CPP due to defects in MKRN3 and DLK1 has not been completely clarified, but it is suggested that both may negatively control the progression of puberty. CPP due to such a single gene abnormality is extremely rare, but it is important to understand the mechanisms of puberty and reproduction. A further development in the genetics of CPP is expected in the future.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
