Alessia Mingarelli, Giovanni Battista Pipitone, Giacomo Torini, Maria Grazia Patricelli, Martina Totaro, Clara Colonna, Paola Carrera, Federico Raviglione
{"title":"Lrp2候选变异体双胞胎(Donnay-Barrow/Foar综合征)的行为表型、电临床特征和治疗选择。","authors":"Alessia Mingarelli, Giovanni Battista Pipitone, Giacomo Torini, Maria Grazia Patricelli, Martina Totaro, Clara Colonna, Paola Carrera, Federico Raviglione","doi":"10.1155/2023/6679572","DOIUrl":null,"url":null,"abstract":"<p><p>The <i>LRP2</i> gene encodes megalin (LRP-2/GP330), a large single-spanning transmembrane glycoprotein that serves as a multiligand endocytotic receptor and mediates the reabsorption of albumin in the proximal renal tubule. <i>LRP2</i> is implicated in an autosomal recessive disorder characterized by dimorphisms, ocular anomalies, sensorineural deafness, proteinuria, epilepsy, and intellectual disability: a clinical condition called Donnai-Barrow syndrome (DBS) or facio-oculo-acoustico-renal (FOAR) syndrome. Pathogenic variants in <i>LRP2</i> have been reported in fewer than 60 patients, but a detailed description of seizures, electroencephalographic patterns, imaging findings, behavioral phenotype, and long-term follow-up is still needed. We provide a clinical report of two mono-chorionic twins with <i>LRP2</i>-related disease manifesting developmental delay, autistic features, seizures, proteinuria, and sleep disorders. By sequencing clinical exome, <i>LRP2</i> candidate rare variants, c.6815G > A, p. (Arg2272His), inherited from the mother and c.12725A > G, p. (Asp4242Gly), inherited from the father, were identified. During follow-up, at the age of 7, the main clinical features of the patients included insomnia, autistic features, severe psychomotor delay, and absent speech. The patients were under treatment with risperidone, antiseizure medications (ASMs), and supplementation of alpha-lactalbumin for self-injury and sleep disturbance. Our study confirmed the wide spectrum of behavioral and neurological and psychiatric features of this rare condition, suggesting new treatment options.</p>","PeriodicalId":30325,"journal":{"name":"Case Reports in Genetics","volume":"2023 ","pages":"6679572"},"PeriodicalIF":0.0000,"publicationDate":"2023-09-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10560113/pdf/","citationCount":"0","resultStr":"{\"title\":\"Behavioral Phenotype, Electroclinical Features, and Treatment Options in Twins with <i>Lrp2</i> Candidate Variants (Donnay-Barrow/Foar Syndrome).\",\"authors\":\"Alessia Mingarelli, Giovanni Battista Pipitone, Giacomo Torini, Maria Grazia Patricelli, Martina Totaro, Clara Colonna, Paola Carrera, Federico Raviglione\",\"doi\":\"10.1155/2023/6679572\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The <i>LRP2</i> gene encodes megalin (LRP-2/GP330), a large single-spanning transmembrane glycoprotein that serves as a multiligand endocytotic receptor and mediates the reabsorption of albumin in the proximal renal tubule. <i>LRP2</i> is implicated in an autosomal recessive disorder characterized by dimorphisms, ocular anomalies, sensorineural deafness, proteinuria, epilepsy, and intellectual disability: a clinical condition called Donnai-Barrow syndrome (DBS) or facio-oculo-acoustico-renal (FOAR) syndrome. Pathogenic variants in <i>LRP2</i> have been reported in fewer than 60 patients, but a detailed description of seizures, electroencephalographic patterns, imaging findings, behavioral phenotype, and long-term follow-up is still needed. We provide a clinical report of two mono-chorionic twins with <i>LRP2</i>-related disease manifesting developmental delay, autistic features, seizures, proteinuria, and sleep disorders. By sequencing clinical exome, <i>LRP2</i> candidate rare variants, c.6815G > A, p. (Arg2272His), inherited from the mother and c.12725A > G, p. (Asp4242Gly), inherited from the father, were identified. During follow-up, at the age of 7, the main clinical features of the patients included insomnia, autistic features, severe psychomotor delay, and absent speech. The patients were under treatment with risperidone, antiseizure medications (ASMs), and supplementation of alpha-lactalbumin for self-injury and sleep disturbance. Our study confirmed the wide spectrum of behavioral and neurological and psychiatric features of this rare condition, suggesting new treatment options.</p>\",\"PeriodicalId\":30325,\"journal\":{\"name\":\"Case Reports in Genetics\",\"volume\":\"2023 \",\"pages\":\"6679572\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-09-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10560113/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Case Reports in Genetics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2023/6679572\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2023/6679572","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Behavioral Phenotype, Electroclinical Features, and Treatment Options in Twins with Lrp2 Candidate Variants (Donnay-Barrow/Foar Syndrome).
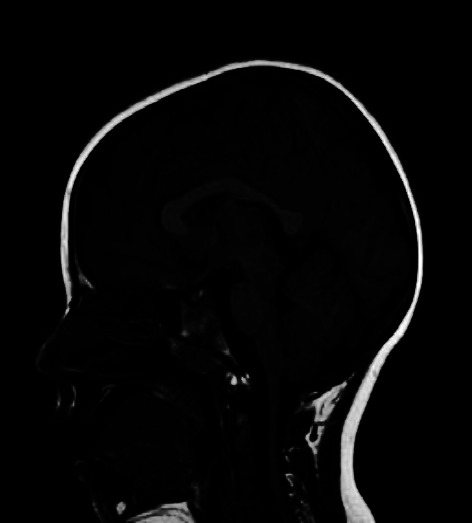
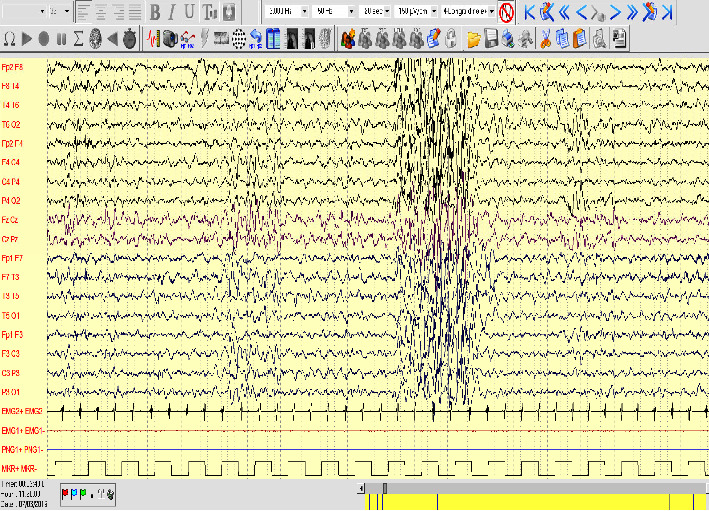
The LRP2 gene encodes megalin (LRP-2/GP330), a large single-spanning transmembrane glycoprotein that serves as a multiligand endocytotic receptor and mediates the reabsorption of albumin in the proximal renal tubule. LRP2 is implicated in an autosomal recessive disorder characterized by dimorphisms, ocular anomalies, sensorineural deafness, proteinuria, epilepsy, and intellectual disability: a clinical condition called Donnai-Barrow syndrome (DBS) or facio-oculo-acoustico-renal (FOAR) syndrome. Pathogenic variants in LRP2 have been reported in fewer than 60 patients, but a detailed description of seizures, electroencephalographic patterns, imaging findings, behavioral phenotype, and long-term follow-up is still needed. We provide a clinical report of two mono-chorionic twins with LRP2-related disease manifesting developmental delay, autistic features, seizures, proteinuria, and sleep disorders. By sequencing clinical exome, LRP2 candidate rare variants, c.6815G > A, p. (Arg2272His), inherited from the mother and c.12725A > G, p. (Asp4242Gly), inherited from the father, were identified. During follow-up, at the age of 7, the main clinical features of the patients included insomnia, autistic features, severe psychomotor delay, and absent speech. The patients were under treatment with risperidone, antiseizure medications (ASMs), and supplementation of alpha-lactalbumin for self-injury and sleep disturbance. Our study confirmed the wide spectrum of behavioral and neurological and psychiatric features of this rare condition, suggesting new treatment options.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
