Hamna Shahid, Nazish Shakoor, Anisa Bibi, Asma Saleem Qazi, Rida Fatima Saeed, Aqeela Nawaz, Sajid Malik, Sara Mumtaz
{"title":"FLNB中的一个停增变异株c.220C>T(p.(Gln74*))在一个血缘家族中分离并伴有强直性脊柱炎综合征。","authors":"Hamna Shahid, Nazish Shakoor, Anisa Bibi, Asma Saleem Qazi, Rida Fatima Saeed, Aqeela Nawaz, Sajid Malik, Sara Mumtaz","doi":"10.59249/UTCP9818","DOIUrl":null,"url":null,"abstract":"<p><p>Spondylocarpotarsal synostosis (SCT) syndrome is a very rare and severe form of skeletal dysplasia. The hallmark features of SCT are disproportionate short stature, scoliosis, fusion of carpal and tarsal bones, and clubfoot. Other common manifestations are cleft palate, conductive and sensorineural hearing loss, joint stiffness, and dental enamel hypoplasia. Homozygous variants in <i>FLNB</i> are known to cause SCT. This study was aimed to investigate the phenotypic and genetic basis of unique presentation of SCT syndrome segregating in a consanguineous Pakistani family. Three of the four affected siblings evaluated had severe short stature, short trunk, short neck, kyphoscoliosis, pectus carinatum, and winged scapula. The subjects had difficulty in walking and gait problems and complained of knee pain and backache. Roentgenographic examination of the eldest patient revealed gross anomalies in the axial skeleton including thoracolumbar and cervical fusion of ribs, severe kyphoscoliosis, thoracic and lumbar lordosis, coxa valga, fusion of certain carpals and tarsals, and clinodactyly. The patients had normal faces and lacked other typical features of SCT like cleft palate, conductive and sensorineural hearing loss, joint stiffness, and dental enamel hypoplasia. Whole exome sequencing (WES) of two affected siblings led to the discovery of a rare stop-gain variant c.220C>T (p.(Gln74*)) in exon 1 of the <i>FLNB</i> gene. The variant was homozygous and segregated with the malformation in this family. This study reports extensive phenotypic variability in SCT and expands the mutation spectrum of <i>FLNB</i>.</p>","PeriodicalId":48617,"journal":{"name":"Yale Journal of Biology and Medicine","volume":"96 3","pages":"383-396"},"PeriodicalIF":3.9000,"publicationDate":"2023-09-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/87/cc/yjbm_96_3_383.PMC10524816.pdf","citationCount":"0","resultStr":"{\"title\":\"A Stop-gain Variant c.220C>T (p.(Gln74*)) in <i>FLNB</i> Segregates with Spondylocarpotarsal Synostosis Syndrome in a Consanguineous Family.\",\"authors\":\"Hamna Shahid, Nazish Shakoor, Anisa Bibi, Asma Saleem Qazi, Rida Fatima Saeed, Aqeela Nawaz, Sajid Malik, Sara Mumtaz\",\"doi\":\"10.59249/UTCP9818\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Spondylocarpotarsal synostosis (SCT) syndrome is a very rare and severe form of skeletal dysplasia. The hallmark features of SCT are disproportionate short stature, scoliosis, fusion of carpal and tarsal bones, and clubfoot. Other common manifestations are cleft palate, conductive and sensorineural hearing loss, joint stiffness, and dental enamel hypoplasia. Homozygous variants in <i>FLNB</i> are known to cause SCT. This study was aimed to investigate the phenotypic and genetic basis of unique presentation of SCT syndrome segregating in a consanguineous Pakistani family. Three of the four affected siblings evaluated had severe short stature, short trunk, short neck, kyphoscoliosis, pectus carinatum, and winged scapula. The subjects had difficulty in walking and gait problems and complained of knee pain and backache. Roentgenographic examination of the eldest patient revealed gross anomalies in the axial skeleton including thoracolumbar and cervical fusion of ribs, severe kyphoscoliosis, thoracic and lumbar lordosis, coxa valga, fusion of certain carpals and tarsals, and clinodactyly. The patients had normal faces and lacked other typical features of SCT like cleft palate, conductive and sensorineural hearing loss, joint stiffness, and dental enamel hypoplasia. Whole exome sequencing (WES) of two affected siblings led to the discovery of a rare stop-gain variant c.220C>T (p.(Gln74*)) in exon 1 of the <i>FLNB</i> gene. The variant was homozygous and segregated with the malformation in this family. This study reports extensive phenotypic variability in SCT and expands the mutation spectrum of <i>FLNB</i>.</p>\",\"PeriodicalId\":48617,\"journal\":{\"name\":\"Yale Journal of Biology and Medicine\",\"volume\":\"96 3\",\"pages\":\"383-396\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2023-09-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/87/cc/yjbm_96_3_383.PMC10524816.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Yale Journal of Biology and Medicine\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://doi.org/10.59249/UTCP9818\",\"RegionNum\":3,\"RegionCategory\":\"工程技术\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/9/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Yale Journal of Biology and Medicine","FirstCategoryId":"5","ListUrlMain":"https://doi.org/10.59249/UTCP9818","RegionNum":3,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"BIOLOGY","Score":null,"Total":0}
A Stop-gain Variant c.220C>T (p.(Gln74*)) in FLNB Segregates with Spondylocarpotarsal Synostosis Syndrome in a Consanguineous Family.
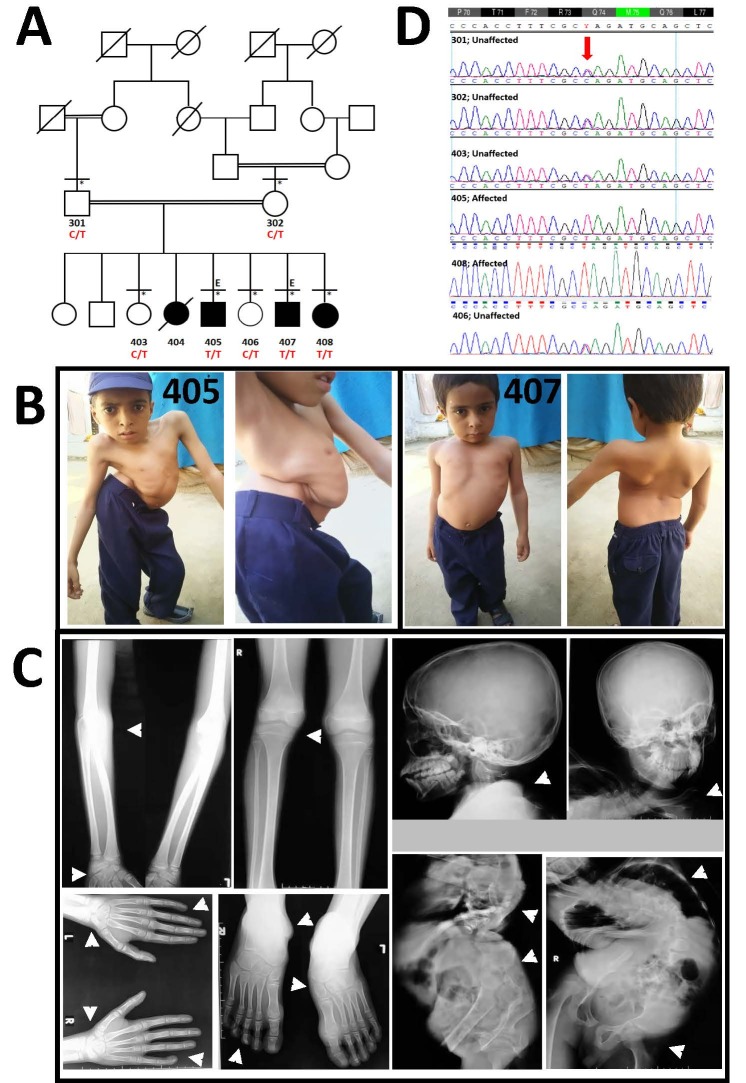
Spondylocarpotarsal synostosis (SCT) syndrome is a very rare and severe form of skeletal dysplasia. The hallmark features of SCT are disproportionate short stature, scoliosis, fusion of carpal and tarsal bones, and clubfoot. Other common manifestations are cleft palate, conductive and sensorineural hearing loss, joint stiffness, and dental enamel hypoplasia. Homozygous variants in FLNB are known to cause SCT. This study was aimed to investigate the phenotypic and genetic basis of unique presentation of SCT syndrome segregating in a consanguineous Pakistani family. Three of the four affected siblings evaluated had severe short stature, short trunk, short neck, kyphoscoliosis, pectus carinatum, and winged scapula. The subjects had difficulty in walking and gait problems and complained of knee pain and backache. Roentgenographic examination of the eldest patient revealed gross anomalies in the axial skeleton including thoracolumbar and cervical fusion of ribs, severe kyphoscoliosis, thoracic and lumbar lordosis, coxa valga, fusion of certain carpals and tarsals, and clinodactyly. The patients had normal faces and lacked other typical features of SCT like cleft palate, conductive and sensorineural hearing loss, joint stiffness, and dental enamel hypoplasia. Whole exome sequencing (WES) of two affected siblings led to the discovery of a rare stop-gain variant c.220C>T (p.(Gln74*)) in exon 1 of the FLNB gene. The variant was homozygous and segregated with the malformation in this family. This study reports extensive phenotypic variability in SCT and expands the mutation spectrum of FLNB.
期刊介绍:
The Yale Journal of Biology and Medicine (YJBM) is a graduate and medical student-run, peer-reviewed, open-access journal dedicated to the publication of original research articles, scientific reviews, articles on medical history, personal perspectives on medicine, policy analyses, case reports, and symposia related to biomedical matters. YJBM is published quarterly and aims to publish articles of interest to both physicians and scientists. YJBM is and has been an internationally distributed journal with a long history of landmark articles. Our contributors feature a notable list of philosophers, statesmen, scientists, and physicians, including Ernst Cassirer, Harvey Cushing, Rene Dubos, Edward Kennedy, Donald Seldin, and Jack Strominger. Our Editorial Board consists of students and faculty members from Yale School of Medicine and Yale University Graduate School of Arts & Sciences. All manuscripts submitted to YJBM are first evaluated on the basis of scientific quality, originality, appropriateness, contribution to the field, and style. Suitable manuscripts are then subject to rigorous, fair, and rapid peer review.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
