Anna Bertolini, Miriam Rigoldi, Annalia Cianflone, Raffaella Mariani, Alberto Piperno, Francesco Canonico, Graziella Cefalo, Francesca Carubbi, Alessandro Simonati, Maria Letizia Urban, Tommaso Beccari, Rossella Parini
{"title":"一组受α-甘露寡糖病影响的意大利患者的长期结果。","authors":"Anna Bertolini, Miriam Rigoldi, Annalia Cianflone, Raffaella Mariani, Alberto Piperno, Francesco Canonico, Graziella Cefalo, Francesca Carubbi, Alessandro Simonati, Maria Letizia Urban, Tommaso Beccari, Rossella Parini","doi":"10.1097/MCD.0000000000000474","DOIUrl":null,"url":null,"abstract":"<p><p>Alpha-mannosidosis (MIM #248500) is an ultra-rare autosomal recessive lysosomal storage disease with multi-system involvement and a wide phenotypic spectrum. Information on long-term outcomes remains poor. We present the long-term outcomes (median, 19 years) of nine patients with alpha-mannosidosis, three females and six males, followed at a single center. The findings of the nine patients were collected from medical records and reported as mean ± SD or median, and range. The age of onset of the first symptoms ranged from 0-1 to 10 years. The diagnostic delay ranged from 2 to 22 years (median= 11 years). Coarse face, hearing, heart valves, joints, gait, language, dysarthria, psychiatric symptoms, I.Q., MRI, walking disabilities, orthopedic disturbances and surgeries showed a slow worsening over the decades. Our patients showed a slowly worsening progressive outcome over the decades. Psychiatric symptoms were present in 100% of our population and improved with the appropriate pharmacological intervention. This aspect requires attention when following up on these patients. Our description of the long-term evolution of alpha-mannosidosis patients may provide basic knowledge for understanding the effects of specific treatments.</p>","PeriodicalId":50682,"journal":{"name":"Clinical Dysmorphology","volume":" ","pages":"1-8"},"PeriodicalIF":0.5000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10702697/pdf/","citationCount":"0","resultStr":"{\"title\":\"Long-term outcome of a cohort of Italian patients affected with alpha-Mannosidosis.\",\"authors\":\"Anna Bertolini, Miriam Rigoldi, Annalia Cianflone, Raffaella Mariani, Alberto Piperno, Francesco Canonico, Graziella Cefalo, Francesca Carubbi, Alessandro Simonati, Maria Letizia Urban, Tommaso Beccari, Rossella Parini\",\"doi\":\"10.1097/MCD.0000000000000474\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Alpha-mannosidosis (MIM #248500) is an ultra-rare autosomal recessive lysosomal storage disease with multi-system involvement and a wide phenotypic spectrum. Information on long-term outcomes remains poor. We present the long-term outcomes (median, 19 years) of nine patients with alpha-mannosidosis, three females and six males, followed at a single center. The findings of the nine patients were collected from medical records and reported as mean ± SD or median, and range. The age of onset of the first symptoms ranged from 0-1 to 10 years. The diagnostic delay ranged from 2 to 22 years (median= 11 years). Coarse face, hearing, heart valves, joints, gait, language, dysarthria, psychiatric symptoms, I.Q., MRI, walking disabilities, orthopedic disturbances and surgeries showed a slow worsening over the decades. Our patients showed a slowly worsening progressive outcome over the decades. Psychiatric symptoms were present in 100% of our population and improved with the appropriate pharmacological intervention. This aspect requires attention when following up on these patients. Our description of the long-term evolution of alpha-mannosidosis patients may provide basic knowledge for understanding the effects of specific treatments.</p>\",\"PeriodicalId\":50682,\"journal\":{\"name\":\"Clinical Dysmorphology\",\"volume\":\" \",\"pages\":\"1-8\"},\"PeriodicalIF\":0.5000,\"publicationDate\":\"2024-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10702697/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Dysmorphology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1097/MCD.0000000000000474\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/11/23 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Dysmorphology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1097/MCD.0000000000000474","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/11/23 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Long-term outcome of a cohort of Italian patients affected with alpha-Mannosidosis.
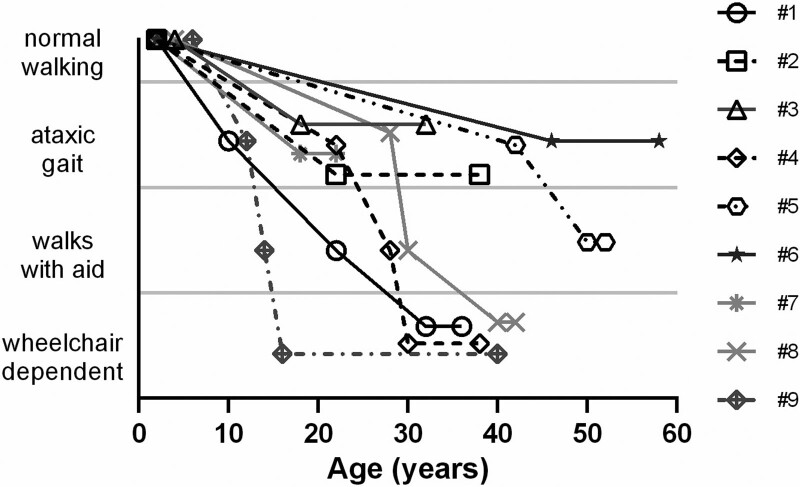
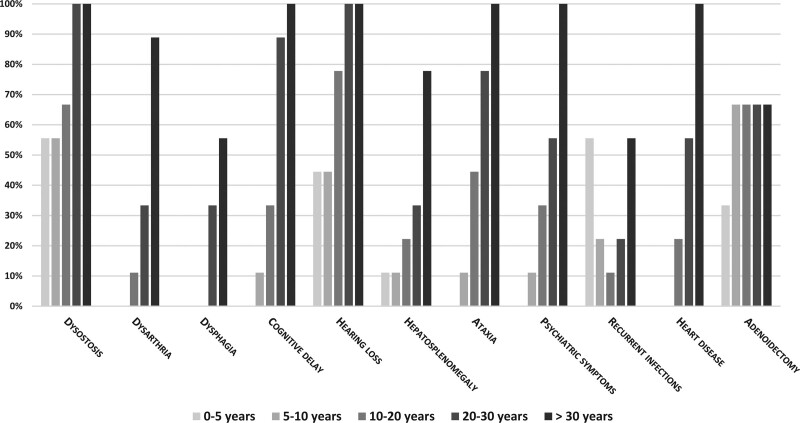
Alpha-mannosidosis (MIM #248500) is an ultra-rare autosomal recessive lysosomal storage disease with multi-system involvement and a wide phenotypic spectrum. Information on long-term outcomes remains poor. We present the long-term outcomes (median, 19 years) of nine patients with alpha-mannosidosis, three females and six males, followed at a single center. The findings of the nine patients were collected from medical records and reported as mean ± SD or median, and range. The age of onset of the first symptoms ranged from 0-1 to 10 years. The diagnostic delay ranged from 2 to 22 years (median= 11 years). Coarse face, hearing, heart valves, joints, gait, language, dysarthria, psychiatric symptoms, I.Q., MRI, walking disabilities, orthopedic disturbances and surgeries showed a slow worsening over the decades. Our patients showed a slowly worsening progressive outcome over the decades. Psychiatric symptoms were present in 100% of our population and improved with the appropriate pharmacological intervention. This aspect requires attention when following up on these patients. Our description of the long-term evolution of alpha-mannosidosis patients may provide basic knowledge for understanding the effects of specific treatments.
期刊介绍:
Clinical Dysmorphology publishes succinct case reports on the etiology, clinical delineation, genetic mapping, and molecular embryology of birth defects. This journal covers such topics as multiple congenital anomaly syndromes - with particular emphasis on previously undescribed conditions, rare findings, ethnic differences in existing syndromes, fetal abnormalities, and cytogenetic aberrations that might give clues to the localization of developmental genes. Regular features include original, peer-reviewed articles, conference reports, book and software reviews, abstracts and summaries from the UK Dysmorphology Club, and literature summaries.
Submitted articles undergo a preliminary review by the editor. Some articles may be returned to authors wihtout further consideration. Those being considered for publication will undergo further assessment and peer-review by the editors and those invited to do so from a reviewer pool.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
