{"title":"肿瘤基因组测序数据的全面而现实的模拟。","authors":"Brian O'Sullivan, Cathal Seoighe","doi":"10.1093/narcan/zcad051","DOIUrl":null,"url":null,"abstract":"<p><p>Accurate identification of somatic mutations and allele frequencies in cancer has critical research and clinical applications. Several computational tools have been developed for this purpose but, in the absence of comprehensive 'ground truth' data, assessing the accuracy of these methods is challenging. We created a computational framework to simulate tumour and matched normal sequencing data for which the source of all loci that contain non-reference bases is known, based on a phased, personalized genome. Unlike existing methods, we account for sampling errors inherent in the sequencing process. Using this framework, we assess accuracy and biases in inferred mutations and their frequencies in an established somatic mutation calling pipeline. We demonstrate bias in existing methods of mutant allele frequency estimation and show, for the first time, the observed mutation frequency spectrum corresponding to a theoretical model of tumour evolution. We highlight the impact of quality filters on detection sensitivity of clinically actionable variants and provide definitive assessment of false positive and false negative mutation calls. Our simulation framework provides an improved means to assess the accuracy of somatic mutation calling pipelines and a detailed picture of the effects of technical parameters and experimental factors on somatic mutation calling in cancer samples.</p>","PeriodicalId":94149,"journal":{"name":"NAR cancer","volume":"5 3","pages":"zcad051"},"PeriodicalIF":3.2000,"publicationDate":"2023-09-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10516706/pdf/","citationCount":"0","resultStr":"{\"title\":\"Comprehensive and realistic simulation of tumour genomic sequencing data.\",\"authors\":\"Brian O'Sullivan, Cathal Seoighe\",\"doi\":\"10.1093/narcan/zcad051\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Accurate identification of somatic mutations and allele frequencies in cancer has critical research and clinical applications. Several computational tools have been developed for this purpose but, in the absence of comprehensive 'ground truth' data, assessing the accuracy of these methods is challenging. We created a computational framework to simulate tumour and matched normal sequencing data for which the source of all loci that contain non-reference bases is known, based on a phased, personalized genome. Unlike existing methods, we account for sampling errors inherent in the sequencing process. Using this framework, we assess accuracy and biases in inferred mutations and their frequencies in an established somatic mutation calling pipeline. We demonstrate bias in existing methods of mutant allele frequency estimation and show, for the first time, the observed mutation frequency spectrum corresponding to a theoretical model of tumour evolution. We highlight the impact of quality filters on detection sensitivity of clinically actionable variants and provide definitive assessment of false positive and false negative mutation calls. Our simulation framework provides an improved means to assess the accuracy of somatic mutation calling pipelines and a detailed picture of the effects of technical parameters and experimental factors on somatic mutation calling in cancer samples.</p>\",\"PeriodicalId\":94149,\"journal\":{\"name\":\"NAR cancer\",\"volume\":\"5 3\",\"pages\":\"zcad051\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2023-09-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10516706/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NAR cancer\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/narcan/zcad051\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/9/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NAR cancer","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/narcan/zcad051","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Comprehensive and realistic simulation of tumour genomic sequencing data.
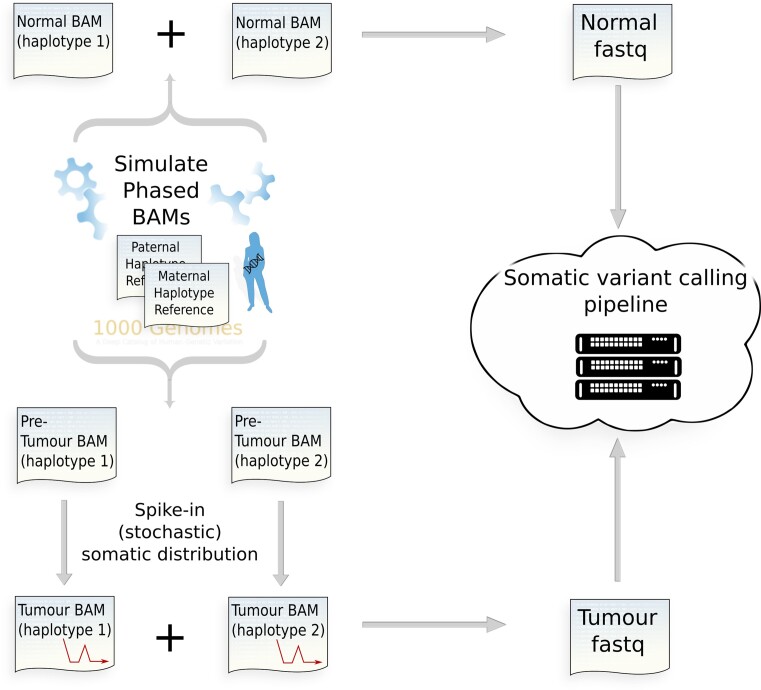
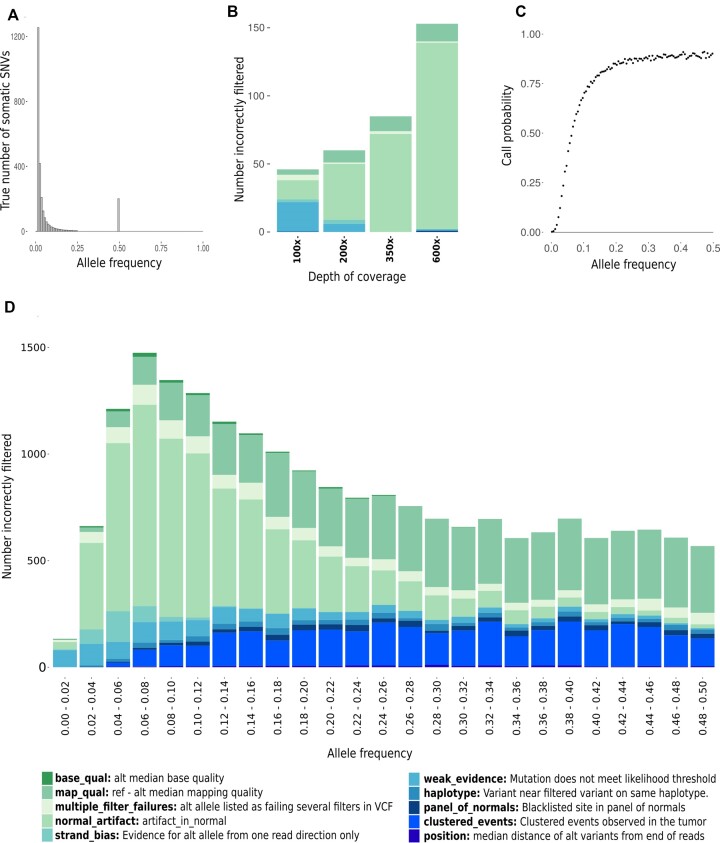
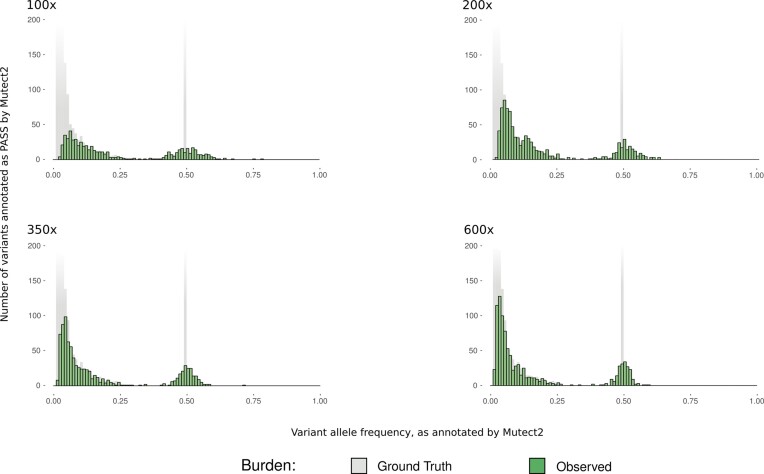
Accurate identification of somatic mutations and allele frequencies in cancer has critical research and clinical applications. Several computational tools have been developed for this purpose but, in the absence of comprehensive 'ground truth' data, assessing the accuracy of these methods is challenging. We created a computational framework to simulate tumour and matched normal sequencing data for which the source of all loci that contain non-reference bases is known, based on a phased, personalized genome. Unlike existing methods, we account for sampling errors inherent in the sequencing process. Using this framework, we assess accuracy and biases in inferred mutations and their frequencies in an established somatic mutation calling pipeline. We demonstrate bias in existing methods of mutant allele frequency estimation and show, for the first time, the observed mutation frequency spectrum corresponding to a theoretical model of tumour evolution. We highlight the impact of quality filters on detection sensitivity of clinically actionable variants and provide definitive assessment of false positive and false negative mutation calls. Our simulation framework provides an improved means to assess the accuracy of somatic mutation calling pipelines and a detailed picture of the effects of technical parameters and experimental factors on somatic mutation calling in cancer samples.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
