Nikola Kresojević, Ivana Perović, Iva Stanković, Aleksandra Tomić, Milica Jecˇmenica Lukic, Vladana Marković, Tanja Stojković, Gorana Mandić, Milena Janković, Ana Marjanović, Marija Branković, Ivana Novaković, Igor Petrović, Nataša Dragašević, Elka Stefanova, Marina Svetel, Vladimir Kostić
{"title":"塞尔维亚共和国亨廷顿舞蹈症患者的临床和遗传特征:单中心经验。","authors":"Nikola Kresojević, Ivana Perović, Iva Stanković, Aleksandra Tomić, Milica Jecˇmenica Lukic, Vladana Marković, Tanja Stojković, Gorana Mandić, Milena Janković, Ana Marjanović, Marija Branković, Ivana Novaković, Igor Petrović, Nataša Dragašević, Elka Stefanova, Marina Svetel, Vladimir Kostić","doi":"10.14802/jmd.23028","DOIUrl":null,"url":null,"abstract":"Dear Editor, Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease caused by an expansion of CAG trinucleotide repeats in the HTT gene, which encodes the huntingtin protein.1 The normal number of CAG repeats in the HTT gene is between 10 and 26; if there are 27–35 repeats, HD will not occur, but due to meiotic instability, the number of repeats may increase, and the disease may manifest in offspring. A total of 36– 39 repeats represent a gray zone with reduced penetrance, where the symptoms occur only in some individuals. Finally, if there are more than 40 CAG repeats, the penetrance is complete, and the disease will certainly manifest.1 Here, we report the clinical features of patients diagnosed with HD at Neurology Clinic, University Clinical Center of Serbia. A previous publication regarding the survival of HD patients in Republic of Serbia was the result of a follow-up study (from 1982 to 2004) in movement disorders department.2 However, the data presented in the current article were collected from an electronic database, which was introduced in Neurology Clinic, University Clinical Center of Serbia in 2003; therefore, some patients may have been included in both studies. A total of 125 patients were found to have sufficient clinical data, including 53 males (42.4%) and 72 females (57.6%). The average age at disease onset (AAO) in our cohort was 45.4 ± 13.3 years (range 13–82 years), similar to the median age of onset of 43 years found in a larger cohort of 1,766 HD patients.3 Juvenile onset is generally rare, occurring in approximately 5% of HD patients with a high number of trinucleotide CAG repeats (over 60).4 In our cohort, only three patients with 64, 71, and 81 CAG repeats had juvenile onset. The small number of juvenile onset patients in our sample may result from referral bias of the tertiary center for adult patients. A total of 16 (14%) patients had a negative family history of HD, a result similar to the rate of 10% reported in the literature and attributed to the anticipation phenomenon in individuals with an intermediate number of CAG repeats (27–35).1,5 In our sample, the most common initial symptom was chorea in 56% of patients, which occurred at a mean age of 47.4 ± 12.5 years (range 14–82 years); 32% had cognitive-psychiatric symptoms (at 41.5 ± 14.0 years, range 13–70 years), and 8.8% had mixed symptoms (at 49.0 ± 14.0 years, range 23–69 years). A similar distribution of initial symptoms was found in a larger multicenter cohort including 1,766 patients, of whom 48% had motor symptoms, 28% had cognitive-psychiatric symptoms (19.6% with psychiatric symptoms and 8.4% with cognitive symptoms), and 13.2% had mixed symptoms.3 We did not find a difference in the number of CAG repeats when comparing the group of patients with psychiatric symptoms at the disease onset","PeriodicalId":16372,"journal":{"name":"Journal of Movement Disorders","volume":"16 3","pages":"333-335"},"PeriodicalIF":2.8000,"publicationDate":"2023-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/3e/2e/jmd-23028.PMC10548084.pdf","citationCount":"0","resultStr":"{\"title\":\"Clinical and Genetic Features of Huntington's Disease Patients From Republic of Serbia: A Single-Center Experience.\",\"authors\":\"Nikola Kresojević, Ivana Perović, Iva Stanković, Aleksandra Tomić, Milica Jecˇmenica Lukic, Vladana Marković, Tanja Stojković, Gorana Mandić, Milena Janković, Ana Marjanović, Marija Branković, Ivana Novaković, Igor Petrović, Nataša Dragašević, Elka Stefanova, Marina Svetel, Vladimir Kostić\",\"doi\":\"10.14802/jmd.23028\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Dear Editor, Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease caused by an expansion of CAG trinucleotide repeats in the HTT gene, which encodes the huntingtin protein.1 The normal number of CAG repeats in the HTT gene is between 10 and 26; if there are 27–35 repeats, HD will not occur, but due to meiotic instability, the number of repeats may increase, and the disease may manifest in offspring. A total of 36– 39 repeats represent a gray zone with reduced penetrance, where the symptoms occur only in some individuals. Finally, if there are more than 40 CAG repeats, the penetrance is complete, and the disease will certainly manifest.1 Here, we report the clinical features of patients diagnosed with HD at Neurology Clinic, University Clinical Center of Serbia. A previous publication regarding the survival of HD patients in Republic of Serbia was the result of a follow-up study (from 1982 to 2004) in movement disorders department.2 However, the data presented in the current article were collected from an electronic database, which was introduced in Neurology Clinic, University Clinical Center of Serbia in 2003; therefore, some patients may have been included in both studies. A total of 125 patients were found to have sufficient clinical data, including 53 males (42.4%) and 72 females (57.6%). The average age at disease onset (AAO) in our cohort was 45.4 ± 13.3 years (range 13–82 years), similar to the median age of onset of 43 years found in a larger cohort of 1,766 HD patients.3 Juvenile onset is generally rare, occurring in approximately 5% of HD patients with a high number of trinucleotide CAG repeats (over 60).4 In our cohort, only three patients with 64, 71, and 81 CAG repeats had juvenile onset. The small number of juvenile onset patients in our sample may result from referral bias of the tertiary center for adult patients. A total of 16 (14%) patients had a negative family history of HD, a result similar to the rate of 10% reported in the literature and attributed to the anticipation phenomenon in individuals with an intermediate number of CAG repeats (27–35).1,5 In our sample, the most common initial symptom was chorea in 56% of patients, which occurred at a mean age of 47.4 ± 12.5 years (range 14–82 years); 32% had cognitive-psychiatric symptoms (at 41.5 ± 14.0 years, range 13–70 years), and 8.8% had mixed symptoms (at 49.0 ± 14.0 years, range 23–69 years). A similar distribution of initial symptoms was found in a larger multicenter cohort including 1,766 patients, of whom 48% had motor symptoms, 28% had cognitive-psychiatric symptoms (19.6% with psychiatric symptoms and 8.4% with cognitive symptoms), and 13.2% had mixed symptoms.3 We did not find a difference in the number of CAG repeats when comparing the group of patients with psychiatric symptoms at the disease onset\",\"PeriodicalId\":16372,\"journal\":{\"name\":\"Journal of Movement Disorders\",\"volume\":\"16 3\",\"pages\":\"333-335\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2023-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/3e/2e/jmd-23028.PMC10548084.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Movement Disorders\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.14802/jmd.23028\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/6/9 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Movement Disorders","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.14802/jmd.23028","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/6/9 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Clinical and Genetic Features of Huntington's Disease Patients From Republic of Serbia: A Single-Center Experience.
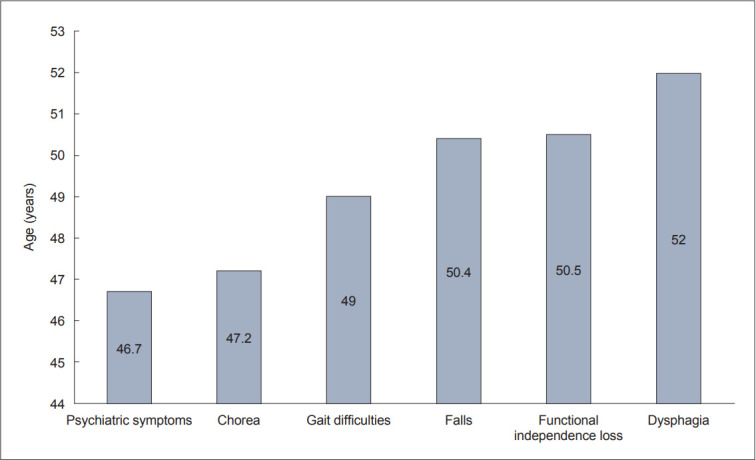
Dear Editor, Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease caused by an expansion of CAG trinucleotide repeats in the HTT gene, which encodes the huntingtin protein.1 The normal number of CAG repeats in the HTT gene is between 10 and 26; if there are 27–35 repeats, HD will not occur, but due to meiotic instability, the number of repeats may increase, and the disease may manifest in offspring. A total of 36– 39 repeats represent a gray zone with reduced penetrance, where the symptoms occur only in some individuals. Finally, if there are more than 40 CAG repeats, the penetrance is complete, and the disease will certainly manifest.1 Here, we report the clinical features of patients diagnosed with HD at Neurology Clinic, University Clinical Center of Serbia. A previous publication regarding the survival of HD patients in Republic of Serbia was the result of a follow-up study (from 1982 to 2004) in movement disorders department.2 However, the data presented in the current article were collected from an electronic database, which was introduced in Neurology Clinic, University Clinical Center of Serbia in 2003; therefore, some patients may have been included in both studies. A total of 125 patients were found to have sufficient clinical data, including 53 males (42.4%) and 72 females (57.6%). The average age at disease onset (AAO) in our cohort was 45.4 ± 13.3 years (range 13–82 years), similar to the median age of onset of 43 years found in a larger cohort of 1,766 HD patients.3 Juvenile onset is generally rare, occurring in approximately 5% of HD patients with a high number of trinucleotide CAG repeats (over 60).4 In our cohort, only three patients with 64, 71, and 81 CAG repeats had juvenile onset. The small number of juvenile onset patients in our sample may result from referral bias of the tertiary center for adult patients. A total of 16 (14%) patients had a negative family history of HD, a result similar to the rate of 10% reported in the literature and attributed to the anticipation phenomenon in individuals with an intermediate number of CAG repeats (27–35).1,5 In our sample, the most common initial symptom was chorea in 56% of patients, which occurred at a mean age of 47.4 ± 12.5 years (range 14–82 years); 32% had cognitive-psychiatric symptoms (at 41.5 ± 14.0 years, range 13–70 years), and 8.8% had mixed symptoms (at 49.0 ± 14.0 years, range 23–69 years). A similar distribution of initial symptoms was found in a larger multicenter cohort including 1,766 patients, of whom 48% had motor symptoms, 28% had cognitive-psychiatric symptoms (19.6% with psychiatric symptoms and 8.4% with cognitive symptoms), and 13.2% had mixed symptoms.3 We did not find a difference in the number of CAG repeats when comparing the group of patients with psychiatric symptoms at the disease onset




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
