Nadine Kirsch-Stefan, Elena Guillen, Niklas Ekman, Sean Barry, Verena Knippel, Sheila Killalea, Martina Weise, Elena Wolff-Holz
{"title":"临床疗效试验的结果在生物仿制药的监管决策中重要吗?","authors":"Nadine Kirsch-Stefan, Elena Guillen, Niklas Ekman, Sean Barry, Verena Knippel, Sheila Killalea, Martina Weise, Elena Wolff-Holz","doi":"10.1007/s40259-023-00631-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>There is an increasing body of evidence supporting a more flexible approach in clinical data requirements for the approval of more complex biosimilar substances such as monoclonal antibodies (mAbs).</p><p><strong>Objective: </strong>The aim of this paper is to further analyse the role of quality/chemistry, manufacturing and controls (CMC) and clinical data for the conclusion on biosimilarity and the decision on marketing authorisation (MA).</p><p><strong>Methods: </strong>In the present study, we analysed the MA applications (MAAs) of all 33 mAbs and three fusion proteins evaluated by the European Medicines Agency (EMA) between July 2012 and November 2022 with special emphasis on all submitted rituximab (four products) and trastuzumab (seven products) biosimilar candidates, including withdrawn applications. For the two withdrawn applications, the comparative efficacy trials suggested biosimilarity, but the quality/CMC package was not accepted by EMA. We therefore investigated whether a negative MAA outcome could have been predicted based on the evidence generated in the quality/CMC packages, regardless of clinical trial data. For this purpose, we reviewed the respective European Public Assessment Reports (EPARs) or withdrawal assessment reports, and the first regulatory assessments for all these 36 MAAs (i.e. day 120 of the centralized procedure), which are not publicly available. During EMA review, where significant issues are identified which would preclude a marketing authorisation, these issues are raised as questions to the applicant and are classified as major objections (MO).</p><p><strong>Results: </strong>In 67% of cases, the outcome of the quality and clinical assessment was the same, i.e. both the quality and clinical assessments either supported approval or did not support approval. In 11% of cases, MO were identified in the quality part of the submission but not in the clinical data. In 22% of cases, MO were raised on the clinical data package but not on the quality data. However, we found no instance where seemingly negative clinical data, including failed efficacy trials, led to a negative overall decision. In each instance, the failure to confirm similar clinical performance in all investigated aspects was eventually viewed as not being related to the biosimilar per se but as being due to imbalances in the trial arms, immaturity of secondary endpoint results, change in the reference product, or even chance findings. Furthermore, when performing an in-depth analysis of the quality and clinical packages of trastuzumab and rituximab biosimilars, we found that in no case were clinical trial data necessary to resolve residual uncertainties regarding the quality part.</p><p><strong>Conclusion: </strong>The results further support the argument that sufficient evidence for biosimilarity can be obtained from a combination of analytical and functional testing and pharmacokinetic studies which may also generate immunogenicity data. This calls into question the usefulness of comparative efficacy studies for the purposes of regulatory decision-making when approving biosimilar mAbs and fusion proteins.</p>","PeriodicalId":9022,"journal":{"name":"BioDrugs","volume":" ","pages":"855-871"},"PeriodicalIF":6.9000,"publicationDate":"2023-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/9b/b1/40259_2023_Article_631.PMC10581956.pdf","citationCount":"2","resultStr":"{\"title\":\"Do the Outcomes of Clinical Efficacy Trials Matter in Regulatory Decision-Making for Biosimilars?\",\"authors\":\"Nadine Kirsch-Stefan, Elena Guillen, Niklas Ekman, Sean Barry, Verena Knippel, Sheila Killalea, Martina Weise, Elena Wolff-Holz\",\"doi\":\"10.1007/s40259-023-00631-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>There is an increasing body of evidence supporting a more flexible approach in clinical data requirements for the approval of more complex biosimilar substances such as monoclonal antibodies (mAbs).</p><p><strong>Objective: </strong>The aim of this paper is to further analyse the role of quality/chemistry, manufacturing and controls (CMC) and clinical data for the conclusion on biosimilarity and the decision on marketing authorisation (MA).</p><p><strong>Methods: </strong>In the present study, we analysed the MA applications (MAAs) of all 33 mAbs and three fusion proteins evaluated by the European Medicines Agency (EMA) between July 2012 and November 2022 with special emphasis on all submitted rituximab (four products) and trastuzumab (seven products) biosimilar candidates, including withdrawn applications. For the two withdrawn applications, the comparative efficacy trials suggested biosimilarity, but the quality/CMC package was not accepted by EMA. We therefore investigated whether a negative MAA outcome could have been predicted based on the evidence generated in the quality/CMC packages, regardless of clinical trial data. For this purpose, we reviewed the respective European Public Assessment Reports (EPARs) or withdrawal assessment reports, and the first regulatory assessments for all these 36 MAAs (i.e. day 120 of the centralized procedure), which are not publicly available. During EMA review, where significant issues are identified which would preclude a marketing authorisation, these issues are raised as questions to the applicant and are classified as major objections (MO).</p><p><strong>Results: </strong>In 67% of cases, the outcome of the quality and clinical assessment was the same, i.e. both the quality and clinical assessments either supported approval or did not support approval. In 11% of cases, MO were identified in the quality part of the submission but not in the clinical data. In 22% of cases, MO were raised on the clinical data package but not on the quality data. However, we found no instance where seemingly negative clinical data, including failed efficacy trials, led to a negative overall decision. In each instance, the failure to confirm similar clinical performance in all investigated aspects was eventually viewed as not being related to the biosimilar per se but as being due to imbalances in the trial arms, immaturity of secondary endpoint results, change in the reference product, or even chance findings. Furthermore, when performing an in-depth analysis of the quality and clinical packages of trastuzumab and rituximab biosimilars, we found that in no case were clinical trial data necessary to resolve residual uncertainties regarding the quality part.</p><p><strong>Conclusion: </strong>The results further support the argument that sufficient evidence for biosimilarity can be obtained from a combination of analytical and functional testing and pharmacokinetic studies which may also generate immunogenicity data. This calls into question the usefulness of comparative efficacy studies for the purposes of regulatory decision-making when approving biosimilar mAbs and fusion proteins.</p>\",\"PeriodicalId\":9022,\"journal\":{\"name\":\"BioDrugs\",\"volume\":\" \",\"pages\":\"855-871\"},\"PeriodicalIF\":6.9000,\"publicationDate\":\"2023-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/9b/b1/40259_2023_Article_631.PMC10581956.pdf\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BioDrugs\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s40259-023-00631-4\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/10/13 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"IMMUNOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BioDrugs","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s40259-023-00631-4","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/10/13 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
Do the Outcomes of Clinical Efficacy Trials Matter in Regulatory Decision-Making for Biosimilars?
Background: There is an increasing body of evidence supporting a more flexible approach in clinical data requirements for the approval of more complex biosimilar substances such as monoclonal antibodies (mAbs).
Objective: The aim of this paper is to further analyse the role of quality/chemistry, manufacturing and controls (CMC) and clinical data for the conclusion on biosimilarity and the decision on marketing authorisation (MA).
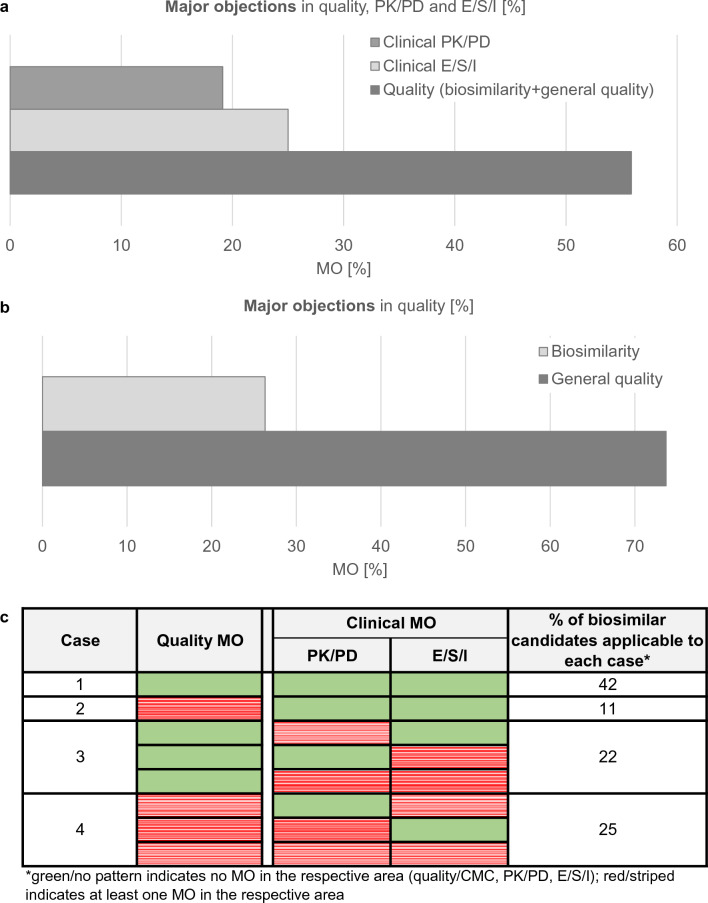
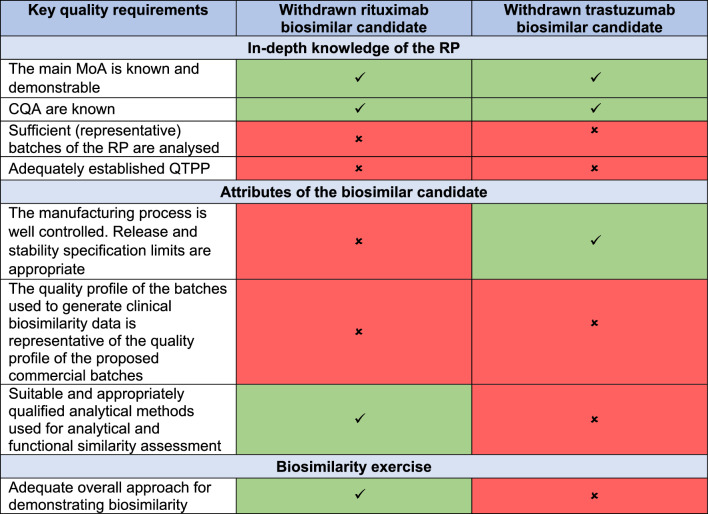
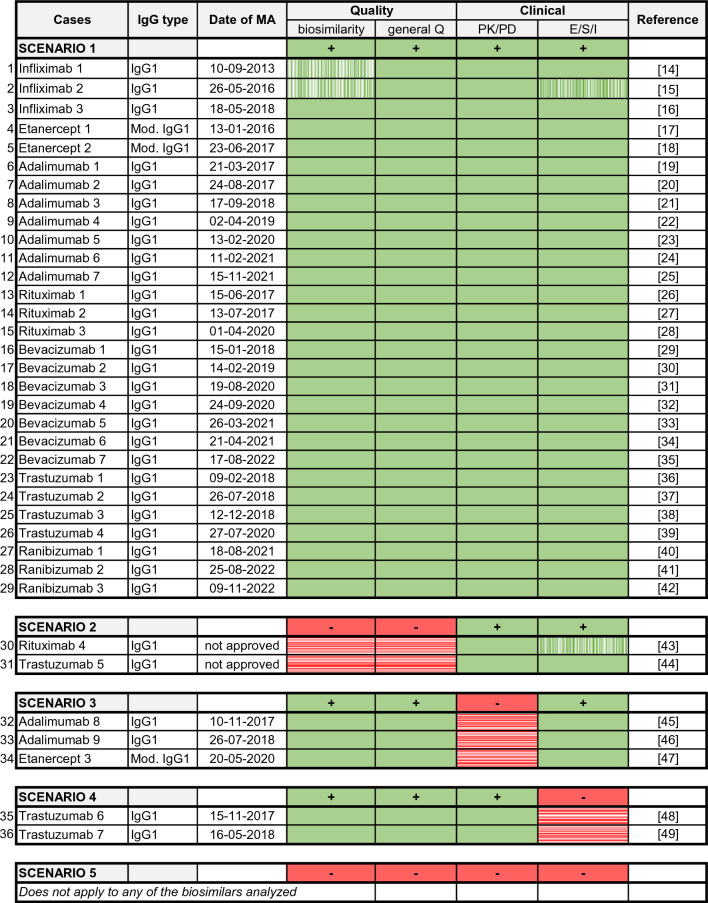
Methods: In the present study, we analysed the MA applications (MAAs) of all 33 mAbs and three fusion proteins evaluated by the European Medicines Agency (EMA) between July 2012 and November 2022 with special emphasis on all submitted rituximab (four products) and trastuzumab (seven products) biosimilar candidates, including withdrawn applications. For the two withdrawn applications, the comparative efficacy trials suggested biosimilarity, but the quality/CMC package was not accepted by EMA. We therefore investigated whether a negative MAA outcome could have been predicted based on the evidence generated in the quality/CMC packages, regardless of clinical trial data. For this purpose, we reviewed the respective European Public Assessment Reports (EPARs) or withdrawal assessment reports, and the first regulatory assessments for all these 36 MAAs (i.e. day 120 of the centralized procedure), which are not publicly available. During EMA review, where significant issues are identified which would preclude a marketing authorisation, these issues are raised as questions to the applicant and are classified as major objections (MO).
Results: In 67% of cases, the outcome of the quality and clinical assessment was the same, i.e. both the quality and clinical assessments either supported approval or did not support approval. In 11% of cases, MO were identified in the quality part of the submission but not in the clinical data. In 22% of cases, MO were raised on the clinical data package but not on the quality data. However, we found no instance where seemingly negative clinical data, including failed efficacy trials, led to a negative overall decision. In each instance, the failure to confirm similar clinical performance in all investigated aspects was eventually viewed as not being related to the biosimilar per se but as being due to imbalances in the trial arms, immaturity of secondary endpoint results, change in the reference product, or even chance findings. Furthermore, when performing an in-depth analysis of the quality and clinical packages of trastuzumab and rituximab biosimilars, we found that in no case were clinical trial data necessary to resolve residual uncertainties regarding the quality part.
Conclusion: The results further support the argument that sufficient evidence for biosimilarity can be obtained from a combination of analytical and functional testing and pharmacokinetic studies which may also generate immunogenicity data. This calls into question the usefulness of comparative efficacy studies for the purposes of regulatory decision-making when approving biosimilar mAbs and fusion proteins.
期刊介绍:
An essential resource for R&D professionals and clinicians with an interest in biologic therapies.
BioDrugs covers the development and therapeutic application of biotechnology-based pharmaceuticals and diagnostic products for the treatment of human disease.
BioDrugs offers a range of additional enhanced features designed to increase the visibility, readership and educational value of the journal’s content. Each article is accompanied by a Key Points summary, giving a time-efficient overview of the content to a wide readership. Articles may be accompanied by plain language summaries to assist patients, caregivers and others in understanding important medical advances. The journal also provides the option to include various other types of enhanced features including slide sets, videos and animations. All enhanced features are peer reviewed to the same high standard as the article itself. Peer review is conducted using Editorial Manager®, supported by a database of international experts. This database is shared with other Adis journals.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
