Sebastian Sten, Tim Cardilin, Madeleine Antonsson, Peter Gennemark
{"title":"N-乙酰氨基半乳糖结合小干扰核糖核酸(GalNAc结合siRNA)的血浆药代动力学。","authors":"Sebastian Sten, Tim Cardilin, Madeleine Antonsson, Peter Gennemark","doi":"10.1007/s40262-023-01314-7","DOIUrl":null,"url":null,"abstract":"<p><p>Small-interfering ribonucleic acids (siRNAs) with N-acetylgalactosamine (GalNAc) conjugation for improved liver uptake represent an emerging class of drugs that modulate liver-expressed therapeutic targets. The pharmacokinetics of GalNAc-siRNAs are characterized by a rapid distribution from plasma to tissue (hours) and a long terminal plasma half-life, analyzed in the form of the antisense strand, driven by redistribution from tissue (weeks). Understanding how clinical pharmacokinetics relate to the dose and type of siRNA chemical stabilizing method used is critical, e.g., to design studies, to investigate safety windows, and to predict the pharmacokinetics of new preclinical assets. To this end, we collected and analyzed pharmacokinetic data from the literature regarding nine GalNAc-siRNAs. Based on this analysis, we showed that the clinical plasma pharmacokinetics of GalNAc-siRNAs are approximately dose proportional and similar between chemical stabilizing methods. This holds for both the area under the concentration-time curve (AUC) and the maximum plasma concentration (C<sub>max</sub>). Corresponding rat and monkey pharmacokinetic data for a subset of the nine GalNAc-siRNAs show dose-proportional C<sub>max</sub>, supra-dose-proportional AUC, and similar pharmacokinetics between chemical stabilizing methods. Together, the animal and human pharmacokinetic data indicate that plasma clearance divided by bioavailability follows allometric principles and scales between species with an exponent of 0.75. Finally, the clinical plasma concentration-time profiles can be empirically described by standard one-compartment kinetics with first-order absorption up to 24 h after subcutaneous dosing, and by three-compartment kinetics with first-order absorption in general. To describe the system more mechanistically, we report a corrected and unambiguously defined version of a previously published physiologically based pharmacokinetic model.</p>","PeriodicalId":10405,"journal":{"name":"Clinical Pharmacokinetics","volume":" ","pages":"1661-1672"},"PeriodicalIF":4.0000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10684612/pdf/","citationCount":"0","resultStr":"{\"title\":\"Plasma Pharmacokinetics of N-Acetylgalactosamine-Conjugated Small-Interfering Ribonucleic Acids (GalNAc-Conjugated siRNAs).\",\"authors\":\"Sebastian Sten, Tim Cardilin, Madeleine Antonsson, Peter Gennemark\",\"doi\":\"10.1007/s40262-023-01314-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Small-interfering ribonucleic acids (siRNAs) with N-acetylgalactosamine (GalNAc) conjugation for improved liver uptake represent an emerging class of drugs that modulate liver-expressed therapeutic targets. The pharmacokinetics of GalNAc-siRNAs are characterized by a rapid distribution from plasma to tissue (hours) and a long terminal plasma half-life, analyzed in the form of the antisense strand, driven by redistribution from tissue (weeks). Understanding how clinical pharmacokinetics relate to the dose and type of siRNA chemical stabilizing method used is critical, e.g., to design studies, to investigate safety windows, and to predict the pharmacokinetics of new preclinical assets. To this end, we collected and analyzed pharmacokinetic data from the literature regarding nine GalNAc-siRNAs. Based on this analysis, we showed that the clinical plasma pharmacokinetics of GalNAc-siRNAs are approximately dose proportional and similar between chemical stabilizing methods. This holds for both the area under the concentration-time curve (AUC) and the maximum plasma concentration (C<sub>max</sub>). Corresponding rat and monkey pharmacokinetic data for a subset of the nine GalNAc-siRNAs show dose-proportional C<sub>max</sub>, supra-dose-proportional AUC, and similar pharmacokinetics between chemical stabilizing methods. Together, the animal and human pharmacokinetic data indicate that plasma clearance divided by bioavailability follows allometric principles and scales between species with an exponent of 0.75. Finally, the clinical plasma concentration-time profiles can be empirically described by standard one-compartment kinetics with first-order absorption up to 24 h after subcutaneous dosing, and by three-compartment kinetics with first-order absorption in general. To describe the system more mechanistically, we report a corrected and unambiguously defined version of a previously published physiologically based pharmacokinetic model.</p>\",\"PeriodicalId\":10405,\"journal\":{\"name\":\"Clinical Pharmacokinetics\",\"volume\":\" \",\"pages\":\"1661-1672\"},\"PeriodicalIF\":4.0000,\"publicationDate\":\"2023-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10684612/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Pharmacokinetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s40262-023-01314-7\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/10/12 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"PHARMACOLOGY & PHARMACY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Pharmacokinetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s40262-023-01314-7","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/10/12 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
Plasma Pharmacokinetics of N-Acetylgalactosamine-Conjugated Small-Interfering Ribonucleic Acids (GalNAc-Conjugated siRNAs).
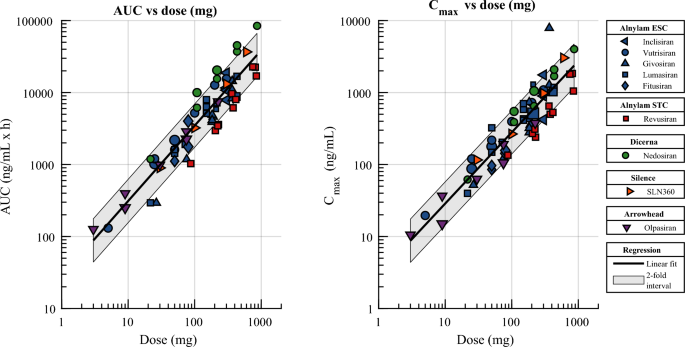
Small-interfering ribonucleic acids (siRNAs) with N-acetylgalactosamine (GalNAc) conjugation for improved liver uptake represent an emerging class of drugs that modulate liver-expressed therapeutic targets. The pharmacokinetics of GalNAc-siRNAs are characterized by a rapid distribution from plasma to tissue (hours) and a long terminal plasma half-life, analyzed in the form of the antisense strand, driven by redistribution from tissue (weeks). Understanding how clinical pharmacokinetics relate to the dose and type of siRNA chemical stabilizing method used is critical, e.g., to design studies, to investigate safety windows, and to predict the pharmacokinetics of new preclinical assets. To this end, we collected and analyzed pharmacokinetic data from the literature regarding nine GalNAc-siRNAs. Based on this analysis, we showed that the clinical plasma pharmacokinetics of GalNAc-siRNAs are approximately dose proportional and similar between chemical stabilizing methods. This holds for both the area under the concentration-time curve (AUC) and the maximum plasma concentration (Cmax). Corresponding rat and monkey pharmacokinetic data for a subset of the nine GalNAc-siRNAs show dose-proportional Cmax, supra-dose-proportional AUC, and similar pharmacokinetics between chemical stabilizing methods. Together, the animal and human pharmacokinetic data indicate that plasma clearance divided by bioavailability follows allometric principles and scales between species with an exponent of 0.75. Finally, the clinical plasma concentration-time profiles can be empirically described by standard one-compartment kinetics with first-order absorption up to 24 h after subcutaneous dosing, and by three-compartment kinetics with first-order absorption in general. To describe the system more mechanistically, we report a corrected and unambiguously defined version of a previously published physiologically based pharmacokinetic model.
期刊介绍:
Clinical Pharmacokinetics promotes the continuing development of clinical pharmacokinetics and pharmacodynamics for the improvement of drug therapy, and for furthering postgraduate education in clinical pharmacology and therapeutics.
Pharmacokinetics, the study of drug disposition in the body, is an integral part of drug development and rational use. Knowledge and application of pharmacokinetic principles leads to accelerated drug development, cost effective drug use and a reduced frequency of adverse effects and drug interactions.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
