Marie-Pierre Sanchez, Thierry Tribout, Naveen K Kadri, Praveen K Chitneedi, Steffen Maak, Chris Hozé, Mekki Boussaha, Pascal Croiseau, Romain Philippe, Mirjam Spengeler, Christa Kühn, Yining Wang, Changxi Li, Graham Plastow, Hubert Pausch, Didier Boichard
{"title":"基于序列的牛肉生产性状GWAS荟萃分析。","authors":"Marie-Pierre Sanchez, Thierry Tribout, Naveen K Kadri, Praveen K Chitneedi, Steffen Maak, Chris Hozé, Mekki Boussaha, Pascal Croiseau, Romain Philippe, Mirjam Spengeler, Christa Kühn, Yining Wang, Changxi Li, Graham Plastow, Hubert Pausch, Didier Boichard","doi":"10.1186/s12711-023-00848-5","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Combining the results of within-population genome-wide association studies (GWAS) based on whole-genome sequences into a single meta-analysis (MA) is an accurate and powerful method for identifying variants associated with complex traits. As part of the H2020 BovReg project, we performed sequence-level MA for beef production traits. Five partners from France, Switzerland, Germany, and Canada contributed summary statistics from sequence-based GWAS conducted with 54,782 animals from 15 purebred or crossbred populations. We combined the summary statistics for four growth, nine morphology, and 15 carcass traits into 16 MA, using both fixed effects and z-score methods.</p><p><strong>Results: </strong>The fixed-effects method was generally more informative to provide indication on potentially causal variants, although we combined substantially different traits in each MA. In comparison with within-population GWAS, this approach highlighted (i) a larger number of quantitative trait loci (QTL), (ii) QTL more frequently located in genomic regions known for their effects on growth and meat/carcass traits, (iii) a smaller number of genomic variants within the QTL, and (iv) candidate variants that were more frequently located in genes. MA pinpointed variants in genes, including MSTN, LCORL, and PLAG1 that have been previously associated with morphology and carcass traits. We also identified dozens of other variants located in genes associated with growth and carcass traits, or with a function that may be related to meat production (e.g., HS6ST1, HERC2, WDR75, COL3A1, SLIT2, MED28, and ANKAR). Some of these variants overlapped with expression or splicing QTL reported in the cattle Genotype-Tissue Expression atlas (CattleGTEx) and could therefore regulate gene expression.</p><p><strong>Conclusions: </strong>By identifying candidate genes and potential causal variants associated with beef production traits in cattle, MA demonstrates great potential for investigating the biological mechanisms underlying these traits. As a complement to within-population GWAS, this approach can provide deeper insights into the genetic architecture of complex traits in beef cattle.</p>","PeriodicalId":55120,"journal":{"name":"Genetics Selection Evolution","volume":"55 1","pages":"70"},"PeriodicalIF":3.1000,"publicationDate":"2023-10-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10568825/pdf/","citationCount":"1","resultStr":"{\"title\":\"Sequence-based GWAS meta-analyses for beef production traits.\",\"authors\":\"Marie-Pierre Sanchez, Thierry Tribout, Naveen K Kadri, Praveen K Chitneedi, Steffen Maak, Chris Hozé, Mekki Boussaha, Pascal Croiseau, Romain Philippe, Mirjam Spengeler, Christa Kühn, Yining Wang, Changxi Li, Graham Plastow, Hubert Pausch, Didier Boichard\",\"doi\":\"10.1186/s12711-023-00848-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Combining the results of within-population genome-wide association studies (GWAS) based on whole-genome sequences into a single meta-analysis (MA) is an accurate and powerful method for identifying variants associated with complex traits. As part of the H2020 BovReg project, we performed sequence-level MA for beef production traits. Five partners from France, Switzerland, Germany, and Canada contributed summary statistics from sequence-based GWAS conducted with 54,782 animals from 15 purebred or crossbred populations. We combined the summary statistics for four growth, nine morphology, and 15 carcass traits into 16 MA, using both fixed effects and z-score methods.</p><p><strong>Results: </strong>The fixed-effects method was generally more informative to provide indication on potentially causal variants, although we combined substantially different traits in each MA. In comparison with within-population GWAS, this approach highlighted (i) a larger number of quantitative trait loci (QTL), (ii) QTL more frequently located in genomic regions known for their effects on growth and meat/carcass traits, (iii) a smaller number of genomic variants within the QTL, and (iv) candidate variants that were more frequently located in genes. MA pinpointed variants in genes, including MSTN, LCORL, and PLAG1 that have been previously associated with morphology and carcass traits. We also identified dozens of other variants located in genes associated with growth and carcass traits, or with a function that may be related to meat production (e.g., HS6ST1, HERC2, WDR75, COL3A1, SLIT2, MED28, and ANKAR). Some of these variants overlapped with expression or splicing QTL reported in the cattle Genotype-Tissue Expression atlas (CattleGTEx) and could therefore regulate gene expression.</p><p><strong>Conclusions: </strong>By identifying candidate genes and potential causal variants associated with beef production traits in cattle, MA demonstrates great potential for investigating the biological mechanisms underlying these traits. As a complement to within-population GWAS, this approach can provide deeper insights into the genetic architecture of complex traits in beef cattle.</p>\",\"PeriodicalId\":55120,\"journal\":{\"name\":\"Genetics Selection Evolution\",\"volume\":\"55 1\",\"pages\":\"70\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2023-10-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10568825/pdf/\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genetics Selection Evolution\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s12711-023-00848-5\",\"RegionNum\":1,\"RegionCategory\":\"农林科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"AGRICULTURE, DAIRY & ANIMAL SCIENCE\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genetics Selection Evolution","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12711-023-00848-5","RegionNum":1,"RegionCategory":"农林科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"AGRICULTURE, DAIRY & ANIMAL SCIENCE","Score":null,"Total":0}
Sequence-based GWAS meta-analyses for beef production traits.
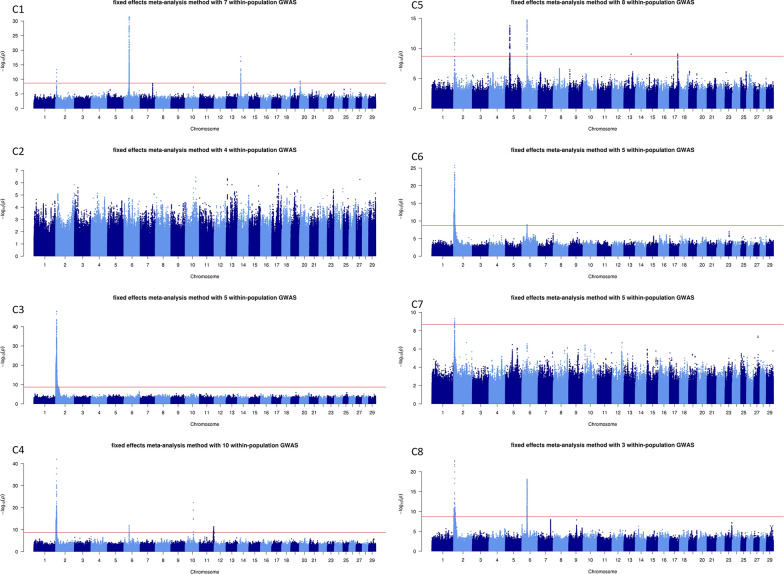
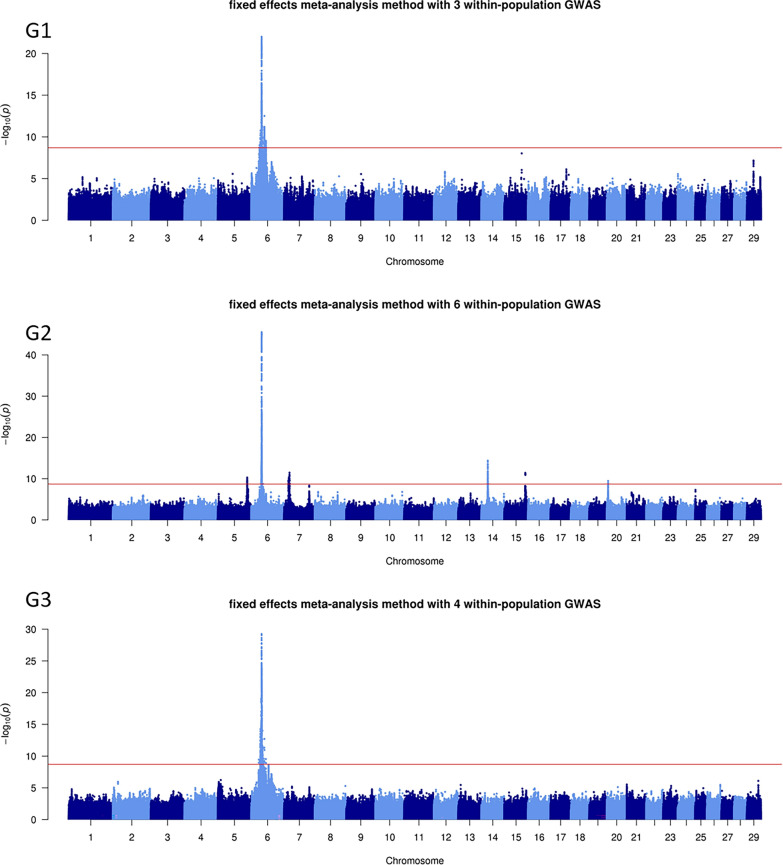
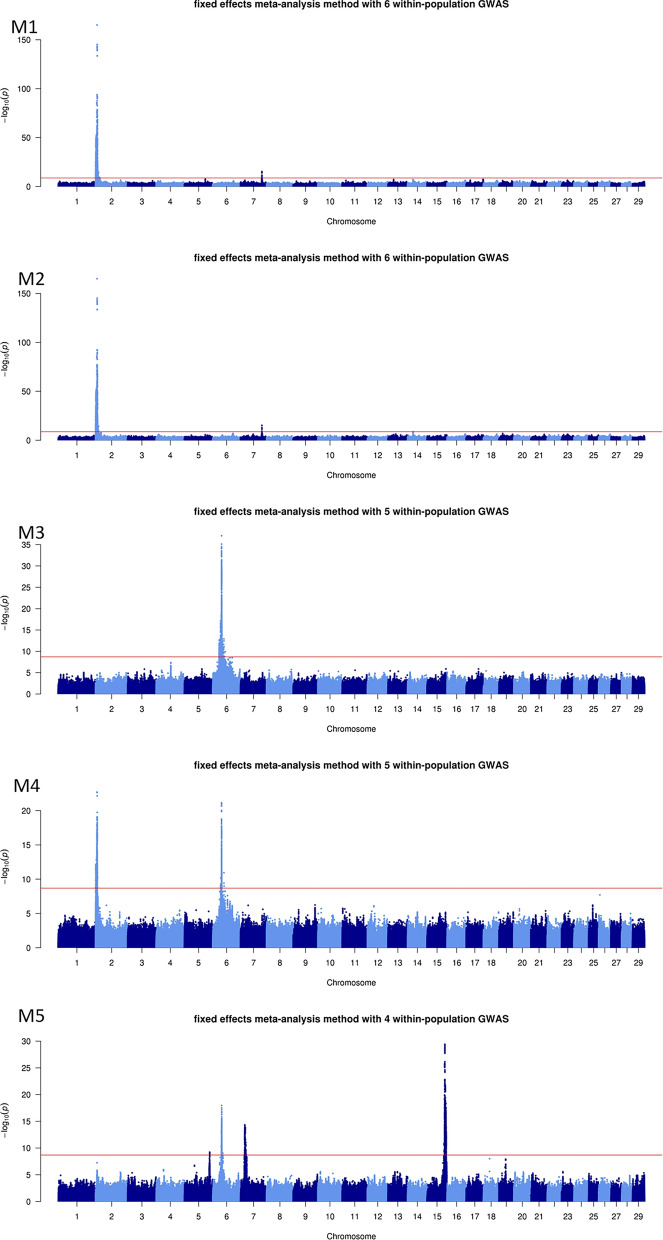
Background: Combining the results of within-population genome-wide association studies (GWAS) based on whole-genome sequences into a single meta-analysis (MA) is an accurate and powerful method for identifying variants associated with complex traits. As part of the H2020 BovReg project, we performed sequence-level MA for beef production traits. Five partners from France, Switzerland, Germany, and Canada contributed summary statistics from sequence-based GWAS conducted with 54,782 animals from 15 purebred or crossbred populations. We combined the summary statistics for four growth, nine morphology, and 15 carcass traits into 16 MA, using both fixed effects and z-score methods.
Results: The fixed-effects method was generally more informative to provide indication on potentially causal variants, although we combined substantially different traits in each MA. In comparison with within-population GWAS, this approach highlighted (i) a larger number of quantitative trait loci (QTL), (ii) QTL more frequently located in genomic regions known for their effects on growth and meat/carcass traits, (iii) a smaller number of genomic variants within the QTL, and (iv) candidate variants that were more frequently located in genes. MA pinpointed variants in genes, including MSTN, LCORL, and PLAG1 that have been previously associated with morphology and carcass traits. We also identified dozens of other variants located in genes associated with growth and carcass traits, or with a function that may be related to meat production (e.g., HS6ST1, HERC2, WDR75, COL3A1, SLIT2, MED28, and ANKAR). Some of these variants overlapped with expression or splicing QTL reported in the cattle Genotype-Tissue Expression atlas (CattleGTEx) and could therefore regulate gene expression.
Conclusions: By identifying candidate genes and potential causal variants associated with beef production traits in cattle, MA demonstrates great potential for investigating the biological mechanisms underlying these traits. As a complement to within-population GWAS, this approach can provide deeper insights into the genetic architecture of complex traits in beef cattle.
期刊介绍:
Genetics Selection Evolution invites basic, applied and methodological content that will aid the current understanding and the utilization of genetic variability in domestic animal species. Although the focus is on domestic animal species, research on other species is invited if it contributes to the understanding of the use of genetic variability in domestic animals. Genetics Selection Evolution publishes results from all levels of study, from the gene to the quantitative trait, from the individual to the population, the breed or the species. Contributions concerning both the biological approach, from molecular genetics to quantitative genetics, as well as the mathematical approach, from population genetics to statistics, are welcome. Specific areas of interest include but are not limited to: gene and QTL identification, mapping and characterization, analysis of new phenotypes, high-throughput SNP data analysis, functional genomics, cytogenetics, genetic diversity of populations and breeds, genetic evaluation, applied and experimental selection, genomic selection, selection efficiency, and statistical methodology for the genetic analysis of phenotypes with quantitative and mixed inheritance.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
