{"title":"沉降速度实验中吉尔伯特理论的自结合模拟:评价最佳拟合模型的指南","authors":"G. R. Bishop, J. J. Correia","doi":"10.1007/s00249-023-01634-3","DOIUrl":null,"url":null,"abstract":"<div><p>There is a long tradition in the Biophysics community of using simulations as a means to understand macromolecular behavior in various physicochemical methods. This allows a rigorous means to interpret observations in terms of fundamental principles, including chemical equilibrium, reaction kinetics, transport processes and thermodynamics. Here we simulate data for the Gilbert Theory for self-association, a fundamental analytical ultracentrifuge (AUC) technique to understand the shape of sedimentation velocity reaction boundaries that involve reversible monomer–Nmer interactions. Simulating monomer–dimer through monomer–hexamer systems as a function of concentration about the equilibrium constant allows a visual means to differentiate reaction stoichiometry by determining end points and inflection positions. Including intermediates (eg A<sub>1</sub>-A<sub>2</sub>-A<sub>3</sub>-A<sub>4</sub>-A<sub>5</sub>-A<sub>6</sub>) in the simulations reveals the smoothing of the reaction boundary and the removal of sharp inflections between monomers and polymers. The addition of cooperativity restores sharp boundaries or peaks to the observation and allows more discrimination in the selection of possible fitting models. Thermodynamic nonideality adds additional features when applied across wide ranges of concentration that might be appropriate for high-concentration therapeutic monoclonal antibody (mAb) solutions. This presentation serves as a tutorial for using modern AUC analysis software like SEDANAL for selecting potential fitting models.</p></div>","PeriodicalId":548,"journal":{"name":"European Biophysics Journal","volume":"52 4-5","pages":"281 - 292"},"PeriodicalIF":2.2000,"publicationDate":"2023-03-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"2","resultStr":"{\"title\":\"Simulation of Gilbert theory for self-association in sedimentation velocity experiments: a guide to evaluate best fitting models\",\"authors\":\"G. R. Bishop, J. J. Correia\",\"doi\":\"10.1007/s00249-023-01634-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>There is a long tradition in the Biophysics community of using simulations as a means to understand macromolecular behavior in various physicochemical methods. This allows a rigorous means to interpret observations in terms of fundamental principles, including chemical equilibrium, reaction kinetics, transport processes and thermodynamics. Here we simulate data for the Gilbert Theory for self-association, a fundamental analytical ultracentrifuge (AUC) technique to understand the shape of sedimentation velocity reaction boundaries that involve reversible monomer–Nmer interactions. Simulating monomer–dimer through monomer–hexamer systems as a function of concentration about the equilibrium constant allows a visual means to differentiate reaction stoichiometry by determining end points and inflection positions. Including intermediates (eg A<sub>1</sub>-A<sub>2</sub>-A<sub>3</sub>-A<sub>4</sub>-A<sub>5</sub>-A<sub>6</sub>) in the simulations reveals the smoothing of the reaction boundary and the removal of sharp inflections between monomers and polymers. The addition of cooperativity restores sharp boundaries or peaks to the observation and allows more discrimination in the selection of possible fitting models. Thermodynamic nonideality adds additional features when applied across wide ranges of concentration that might be appropriate for high-concentration therapeutic monoclonal antibody (mAb) solutions. This presentation serves as a tutorial for using modern AUC analysis software like SEDANAL for selecting potential fitting models.</p></div>\",\"PeriodicalId\":548,\"journal\":{\"name\":\"European Biophysics Journal\",\"volume\":\"52 4-5\",\"pages\":\"281 - 292\"},\"PeriodicalIF\":2.2000,\"publicationDate\":\"2023-03-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"European Biophysics Journal\",\"FirstCategoryId\":\"2\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00249-023-01634-3\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOPHYSICS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Biophysics Journal","FirstCategoryId":"2","ListUrlMain":"https://link.springer.com/article/10.1007/s00249-023-01634-3","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOPHYSICS","Score":null,"Total":0}
Simulation of Gilbert theory for self-association in sedimentation velocity experiments: a guide to evaluate best fitting models
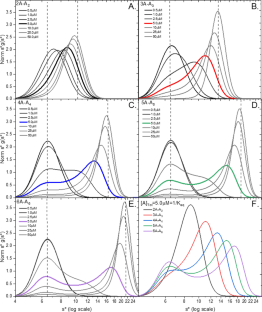
There is a long tradition in the Biophysics community of using simulations as a means to understand macromolecular behavior in various physicochemical methods. This allows a rigorous means to interpret observations in terms of fundamental principles, including chemical equilibrium, reaction kinetics, transport processes and thermodynamics. Here we simulate data for the Gilbert Theory for self-association, a fundamental analytical ultracentrifuge (AUC) technique to understand the shape of sedimentation velocity reaction boundaries that involve reversible monomer–Nmer interactions. Simulating monomer–dimer through monomer–hexamer systems as a function of concentration about the equilibrium constant allows a visual means to differentiate reaction stoichiometry by determining end points and inflection positions. Including intermediates (eg A1-A2-A3-A4-A5-A6) in the simulations reveals the smoothing of the reaction boundary and the removal of sharp inflections between monomers and polymers. The addition of cooperativity restores sharp boundaries or peaks to the observation and allows more discrimination in the selection of possible fitting models. Thermodynamic nonideality adds additional features when applied across wide ranges of concentration that might be appropriate for high-concentration therapeutic monoclonal antibody (mAb) solutions. This presentation serves as a tutorial for using modern AUC analysis software like SEDANAL for selecting potential fitting models.
期刊介绍:
The journal publishes papers in the field of biophysics, which is defined as the study of biological phenomena by using physical methods and concepts. Original papers, reviews and Biophysics letters are published. The primary goal of this journal is to advance the understanding of biological structure and function by application of the principles of physical science, and by presenting the work in a biophysical context.
Papers employing a distinctively biophysical approach at all levels of biological organisation will be considered, as will both experimental and theoretical studies. The criteria for acceptance are scientific content, originality and relevance to biological systems of current interest and importance.
Principal areas of interest include:
- Structure and dynamics of biological macromolecules
- Membrane biophysics and ion channels
- Cell biophysics and organisation
- Macromolecular assemblies
- Biophysical methods and instrumentation
- Advanced microscopics
- System dynamics.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
