{"title":"无偏的,可扩展的采样的蛋白质环构象的概率先验","authors":"Yajia Zhang, Kris Hauser","doi":"10.1186/1472-6807-13-S1-S9","DOIUrl":null,"url":null,"abstract":"<p>Protein loops are flexible structures that are intimately tied to function, but understanding loop motion and generating loop conformation ensembles remain significant computational challenges. Discrete search techniques scale poorly to large loops, optimization and molecular dynamics techniques are prone to local minima, and inverse kinematics techniques can only incorporate structural preferences in adhoc fashion. This paper presents Sub-Loop Inverse Kinematics Monte Carlo (SLIKMC), a new Markov chain Monte Carlo algorithm for generating conformations of closed loops according to experimentally available, heterogeneous structural preferences.</p><p>Our simulation experiments demonstrate that the method computes high-scoring conformations of large loops (<i>></i> 10 residues) orders of magnitude faster than standard Monte Carlo and discrete search techniques. Two new developments contribute to the scalability of the new method. First, structural preferences are specified via a probabilistic graphical model (PGM) that links conformation variables, spatial variables (e.g., atom positions), constraints and prior information in a unified framework. The method uses a sparse PGM that exploits locality of interactions between atoms and residues. Second, a novel method for sampling sub-loops is developed to generate statistically unbiased samples of probability densities restricted by loop-closure constraints.</p><p>Numerical experiments confirm that SLIKMC generates conformation ensembles that are statistically consistent with specified structural preferences. Protein conformations with 100+ residues are sampled on standard PC hardware in seconds. Application to proteins involved in ion-binding demonstrate its potential as a tool for loop ensemble generation and missing structure completion.</p>","PeriodicalId":51240,"journal":{"name":"BMC Structural Biology","volume":"13 1","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2013-11-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1472-6807-13-S1-S9","citationCount":"7","resultStr":"{\"title\":\"Unbiased, scalable sampling of protein loop conformations from probabilistic priors\",\"authors\":\"Yajia Zhang, Kris Hauser\",\"doi\":\"10.1186/1472-6807-13-S1-S9\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Protein loops are flexible structures that are intimately tied to function, but understanding loop motion and generating loop conformation ensembles remain significant computational challenges. Discrete search techniques scale poorly to large loops, optimization and molecular dynamics techniques are prone to local minima, and inverse kinematics techniques can only incorporate structural preferences in adhoc fashion. This paper presents Sub-Loop Inverse Kinematics Monte Carlo (SLIKMC), a new Markov chain Monte Carlo algorithm for generating conformations of closed loops according to experimentally available, heterogeneous structural preferences.</p><p>Our simulation experiments demonstrate that the method computes high-scoring conformations of large loops (<i>></i> 10 residues) orders of magnitude faster than standard Monte Carlo and discrete search techniques. Two new developments contribute to the scalability of the new method. First, structural preferences are specified via a probabilistic graphical model (PGM) that links conformation variables, spatial variables (e.g., atom positions), constraints and prior information in a unified framework. The method uses a sparse PGM that exploits locality of interactions between atoms and residues. Second, a novel method for sampling sub-loops is developed to generate statistically unbiased samples of probability densities restricted by loop-closure constraints.</p><p>Numerical experiments confirm that SLIKMC generates conformation ensembles that are statistically consistent with specified structural preferences. Protein conformations with 100+ residues are sampled on standard PC hardware in seconds. Application to proteins involved in ion-binding demonstrate its potential as a tool for loop ensemble generation and missing structure completion.</p>\",\"PeriodicalId\":51240,\"journal\":{\"name\":\"BMC Structural Biology\",\"volume\":\"13 1\",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2013-11-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/1472-6807-13-S1-S9\",\"citationCount\":\"7\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Structural Biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://link.springer.com/article/10.1186/1472-6807-13-S1-S9\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Structural Biology","FirstCategoryId":"1085","ListUrlMain":"https://link.springer.com/article/10.1186/1472-6807-13-S1-S9","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Unbiased, scalable sampling of protein loop conformations from probabilistic priors
Protein loops are flexible structures that are intimately tied to function, but understanding loop motion and generating loop conformation ensembles remain significant computational challenges. Discrete search techniques scale poorly to large loops, optimization and molecular dynamics techniques are prone to local minima, and inverse kinematics techniques can only incorporate structural preferences in adhoc fashion. This paper presents Sub-Loop Inverse Kinematics Monte Carlo (SLIKMC), a new Markov chain Monte Carlo algorithm for generating conformations of closed loops according to experimentally available, heterogeneous structural preferences.
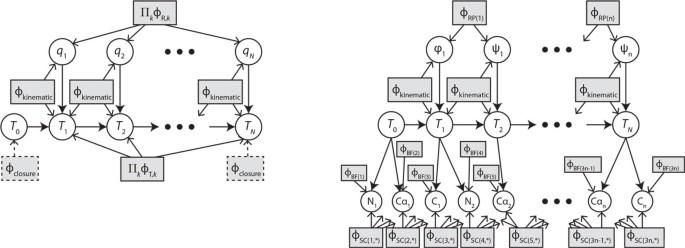
Our simulation experiments demonstrate that the method computes high-scoring conformations of large loops (> 10 residues) orders of magnitude faster than standard Monte Carlo and discrete search techniques. Two new developments contribute to the scalability of the new method. First, structural preferences are specified via a probabilistic graphical model (PGM) that links conformation variables, spatial variables (e.g., atom positions), constraints and prior information in a unified framework. The method uses a sparse PGM that exploits locality of interactions between atoms and residues. Second, a novel method for sampling sub-loops is developed to generate statistically unbiased samples of probability densities restricted by loop-closure constraints.
Numerical experiments confirm that SLIKMC generates conformation ensembles that are statistically consistent with specified structural preferences. Protein conformations with 100+ residues are sampled on standard PC hardware in seconds. Application to proteins involved in ion-binding demonstrate its potential as a tool for loop ensemble generation and missing structure completion.
期刊介绍:
BMC Structural Biology is an open access, peer-reviewed journal that considers articles on investigations into the structure of biological macromolecules, including solving structures, structural and functional analyses, and computational modeling.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
