{"title":"OH + ch3oh吸氢反应的全维势能面理论动力学分析","authors":"Joaquín Espinosa-Garcia, Moisés Garcia-Chamorro","doi":"10.1002/kin.21653","DOIUrl":null,"url":null,"abstract":"<p>Based on an analytical full-dimensional potential energy surface (PES), named PES-2022, fitted to high-level ab initio calculations previously developed by our group and specifically developed to describe this polyatomic reactive process, an exhaustive kinetics analysis was performed in the temperature range 50–2000 K, that is, interstellar, atmospheric and combustion conditions. Using the competitive canonical unified theory with multidimensional tunneling corrections of small curvature, CCUS/SCT, and low- and high-pressure limit (LPL and HPL) models, in this wide temperature range we found that the overall rate constants increase with temperature at T > 300 K and T < 200 K, showing a V-shaped temperature dependence, reproducing the experimental evidence when the HPL model was used. The increase of the rate constant with temperature at low temperatures was due to the strong contribution of the tunneling factor. The title reaction evolves by two paths, H<sub>2</sub>O + CH<sub>2</sub>OH (R1) and H<sub>2</sub>O + CH<sub>3</sub>O (R2), and the branching ratio analysis showed that the R2 path was dominant at T < 200 K while the R1 path dominated at T > 300 K, with a turnover temperature of ∼260 K, in agreement with previous theoretical estimations. Three kinetics isotope effects (KIEs), <sup>13</sup>CH<sub>3</sub>OH, CH<sub>3</sub><sup>18</sup>OH, and CD<sub>3</sub>OH, were theoretically studied, reproducing the experimental evidence. The kinetics analysis in the present paper together with the dynamics study previously reported showed the capacity of the PES-2022 to understand this important chemical process.</p>","PeriodicalId":13894,"journal":{"name":"International Journal of Chemical Kinetics","volume":"55 9","pages":"525-536"},"PeriodicalIF":1.6000,"publicationDate":"2023-05-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/kin.21653","citationCount":"0","resultStr":"{\"title\":\"Theoretical kinetics analysis of the OH + CH3OH hydrogen abstraction reaction using a full-dimensional potential energy surface\",\"authors\":\"Joaquín Espinosa-Garcia, Moisés Garcia-Chamorro\",\"doi\":\"10.1002/kin.21653\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Based on an analytical full-dimensional potential energy surface (PES), named PES-2022, fitted to high-level ab initio calculations previously developed by our group and specifically developed to describe this polyatomic reactive process, an exhaustive kinetics analysis was performed in the temperature range 50–2000 K, that is, interstellar, atmospheric and combustion conditions. Using the competitive canonical unified theory with multidimensional tunneling corrections of small curvature, CCUS/SCT, and low- and high-pressure limit (LPL and HPL) models, in this wide temperature range we found that the overall rate constants increase with temperature at T > 300 K and T < 200 K, showing a V-shaped temperature dependence, reproducing the experimental evidence when the HPL model was used. The increase of the rate constant with temperature at low temperatures was due to the strong contribution of the tunneling factor. The title reaction evolves by two paths, H<sub>2</sub>O + CH<sub>2</sub>OH (R1) and H<sub>2</sub>O + CH<sub>3</sub>O (R2), and the branching ratio analysis showed that the R2 path was dominant at T < 200 K while the R1 path dominated at T > 300 K, with a turnover temperature of ∼260 K, in agreement with previous theoretical estimations. Three kinetics isotope effects (KIEs), <sup>13</sup>CH<sub>3</sub>OH, CH<sub>3</sub><sup>18</sup>OH, and CD<sub>3</sub>OH, were theoretically studied, reproducing the experimental evidence. The kinetics analysis in the present paper together with the dynamics study previously reported showed the capacity of the PES-2022 to understand this important chemical process.</p>\",\"PeriodicalId\":13894,\"journal\":{\"name\":\"International Journal of Chemical Kinetics\",\"volume\":\"55 9\",\"pages\":\"525-536\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2023-05-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/kin.21653\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International Journal of Chemical Kinetics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/kin.21653\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Chemical Kinetics","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/kin.21653","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Theoretical kinetics analysis of the OH + CH3OH hydrogen abstraction reaction using a full-dimensional potential energy surface
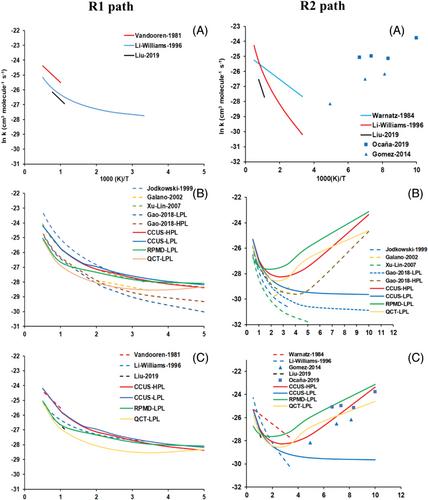
Based on an analytical full-dimensional potential energy surface (PES), named PES-2022, fitted to high-level ab initio calculations previously developed by our group and specifically developed to describe this polyatomic reactive process, an exhaustive kinetics analysis was performed in the temperature range 50–2000 K, that is, interstellar, atmospheric and combustion conditions. Using the competitive canonical unified theory with multidimensional tunneling corrections of small curvature, CCUS/SCT, and low- and high-pressure limit (LPL and HPL) models, in this wide temperature range we found that the overall rate constants increase with temperature at T > 300 K and T < 200 K, showing a V-shaped temperature dependence, reproducing the experimental evidence when the HPL model was used. The increase of the rate constant with temperature at low temperatures was due to the strong contribution of the tunneling factor. The title reaction evolves by two paths, H2O + CH2OH (R1) and H2O + CH3O (R2), and the branching ratio analysis showed that the R2 path was dominant at T < 200 K while the R1 path dominated at T > 300 K, with a turnover temperature of ∼260 K, in agreement with previous theoretical estimations. Three kinetics isotope effects (KIEs), 13CH3OH, CH318OH, and CD3OH, were theoretically studied, reproducing the experimental evidence. The kinetics analysis in the present paper together with the dynamics study previously reported showed the capacity of the PES-2022 to understand this important chemical process.
期刊介绍:
As the leading archival journal devoted exclusively to chemical kinetics, the International Journal of Chemical Kinetics publishes original research in gas phase, condensed phase, and polymer reaction kinetics, as well as biochemical and surface kinetics. The Journal seeks to be the primary archive for careful experimental measurements of reaction kinetics, in both simple and complex systems. The Journal also presents new developments in applied theoretical kinetics and publishes large kinetic models, and the algorithms and estimates used in these models. These include methods for handling the large reaction networks important in biochemistry, catalysis, and free radical chemistry. In addition, the Journal explores such topics as the quantitative relationships between molecular structure and chemical reactivity, organic/inorganic chemistry and reaction mechanisms, and the reactive chemistry at interfaces.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
