Janosch Hennig, Sjoerd J de Vries, Klaus DM Hennig, Leah Randles, Kylie J Walters, Maria Sunnerhagen, Alexandre MJJ Bonvin
{"title":"MTMDAT-HADDOCK:基于有限蛋白水解和质谱的高通量蛋白质复合物结构建模","authors":"Janosch Hennig, Sjoerd J de Vries, Klaus DM Hennig, Leah Randles, Kylie J Walters, Maria Sunnerhagen, Alexandre MJJ Bonvin","doi":"10.1186/1472-6807-12-29","DOIUrl":null,"url":null,"abstract":"<p>MTMDAT is a program designed to facilitate analysis of mass spectrometry data of proteins and biomolecular complexes that are probed structurally by limited proteolysis. This approach can provide information about stable fragments of multidomain proteins, yield tertiary and quaternary structure data, and help determine the origin of stability changes at the amino acid residue level. Here, we introduce a pipeline between MTMDAT and HADDOCK, that facilitates protein-protein complex structure probing in a high-throughput and highly automated fashion.</p><p>A new feature of MTMDAT allows for the direct identification of residues that are involved in complex formation by comparing the mass spectra of bound and unbound proteins after proteolysis. If 3D structures of the unbound components are available, this data can be used to define restraints for data-driven docking to calculate a model of the complex. We describe here a new implementation of MTMDAT, which includes a pipeline to the data-driven docking program HADDOCK, thus streamlining the entire procedure. This addition, together with usability improvements in MTMDAT, enables high-throughput modeling of protein complexes from mass spectrometry data. The algorithm has been validated by using the protein-protein interaction between the ubiquitin-binding domain of proteasome component Rpn13 and ubiquitin. The resulting structural model, based on restraints extracted by MTMDAT from limited proteolysis and modeled by HADDOCK, was compared to the published NMR structure, which relied on twelve unambiguous intermolecular NOE interactions. The MTMDAT-HADDOCK structure was of similar quality to structures generated using only chemical shift perturbation data derived by NMR titration experiments.</p><p>The new MTMDAT-HADDOCK pipeline enables direct high-throughput modeling of protein complexes from mass spectrometry data. MTMDAT-HADDOCK can be downloaded from http://www.ifm.liu.se/chemistry/molbiotech/maria_sunnerhagens_group/mtmdat/together with the manual and example files. The program is free for academic/non-commercial purposes.</p>","PeriodicalId":51240,"journal":{"name":"BMC Structural Biology","volume":"12 1","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2012-11-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1472-6807-12-29","citationCount":"12","resultStr":"{\"title\":\"MTMDAT-HADDOCK: High-throughput, protein complex structure modeling based on limited proteolysis and mass spectrometry\",\"authors\":\"Janosch Hennig, Sjoerd J de Vries, Klaus DM Hennig, Leah Randles, Kylie J Walters, Maria Sunnerhagen, Alexandre MJJ Bonvin\",\"doi\":\"10.1186/1472-6807-12-29\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>MTMDAT is a program designed to facilitate analysis of mass spectrometry data of proteins and biomolecular complexes that are probed structurally by limited proteolysis. This approach can provide information about stable fragments of multidomain proteins, yield tertiary and quaternary structure data, and help determine the origin of stability changes at the amino acid residue level. Here, we introduce a pipeline between MTMDAT and HADDOCK, that facilitates protein-protein complex structure probing in a high-throughput and highly automated fashion.</p><p>A new feature of MTMDAT allows for the direct identification of residues that are involved in complex formation by comparing the mass spectra of bound and unbound proteins after proteolysis. If 3D structures of the unbound components are available, this data can be used to define restraints for data-driven docking to calculate a model of the complex. We describe here a new implementation of MTMDAT, which includes a pipeline to the data-driven docking program HADDOCK, thus streamlining the entire procedure. This addition, together with usability improvements in MTMDAT, enables high-throughput modeling of protein complexes from mass spectrometry data. The algorithm has been validated by using the protein-protein interaction between the ubiquitin-binding domain of proteasome component Rpn13 and ubiquitin. The resulting structural model, based on restraints extracted by MTMDAT from limited proteolysis and modeled by HADDOCK, was compared to the published NMR structure, which relied on twelve unambiguous intermolecular NOE interactions. The MTMDAT-HADDOCK structure was of similar quality to structures generated using only chemical shift perturbation data derived by NMR titration experiments.</p><p>The new MTMDAT-HADDOCK pipeline enables direct high-throughput modeling of protein complexes from mass spectrometry data. MTMDAT-HADDOCK can be downloaded from http://www.ifm.liu.se/chemistry/molbiotech/maria_sunnerhagens_group/mtmdat/together with the manual and example files. The program is free for academic/non-commercial purposes.</p>\",\"PeriodicalId\":51240,\"journal\":{\"name\":\"BMC Structural Biology\",\"volume\":\"12 1\",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2012-11-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/1472-6807-12-29\",\"citationCount\":\"12\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Structural Biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://link.springer.com/article/10.1186/1472-6807-12-29\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Structural Biology","FirstCategoryId":"1085","ListUrlMain":"https://link.springer.com/article/10.1186/1472-6807-12-29","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
MTMDAT-HADDOCK: High-throughput, protein complex structure modeling based on limited proteolysis and mass spectrometry
MTMDAT is a program designed to facilitate analysis of mass spectrometry data of proteins and biomolecular complexes that are probed structurally by limited proteolysis. This approach can provide information about stable fragments of multidomain proteins, yield tertiary and quaternary structure data, and help determine the origin of stability changes at the amino acid residue level. Here, we introduce a pipeline between MTMDAT and HADDOCK, that facilitates protein-protein complex structure probing in a high-throughput and highly automated fashion.
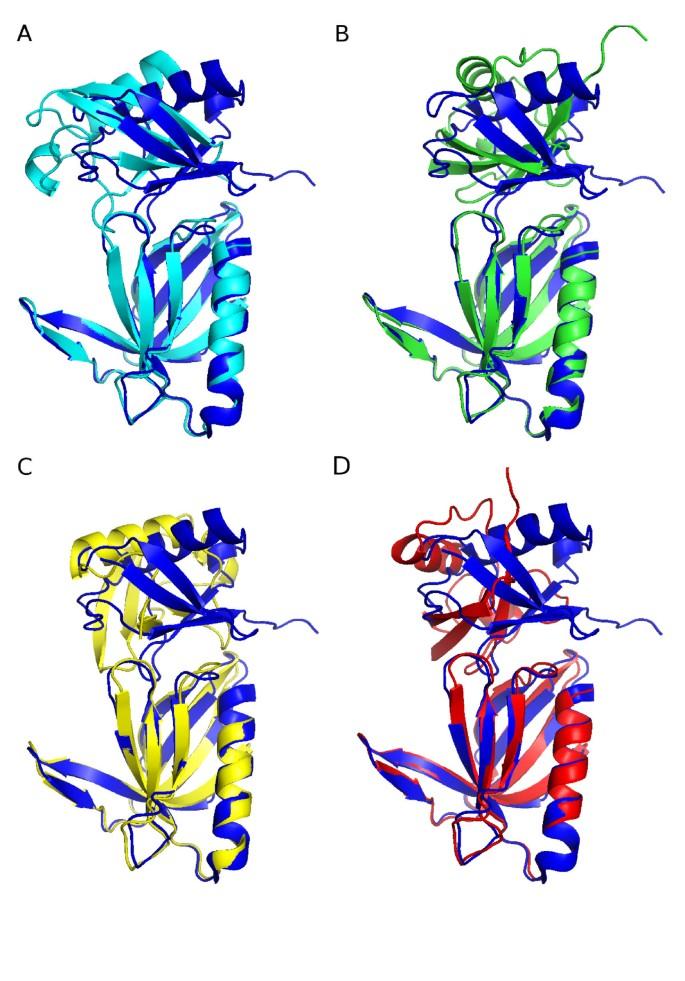
A new feature of MTMDAT allows for the direct identification of residues that are involved in complex formation by comparing the mass spectra of bound and unbound proteins after proteolysis. If 3D structures of the unbound components are available, this data can be used to define restraints for data-driven docking to calculate a model of the complex. We describe here a new implementation of MTMDAT, which includes a pipeline to the data-driven docking program HADDOCK, thus streamlining the entire procedure. This addition, together with usability improvements in MTMDAT, enables high-throughput modeling of protein complexes from mass spectrometry data. The algorithm has been validated by using the protein-protein interaction between the ubiquitin-binding domain of proteasome component Rpn13 and ubiquitin. The resulting structural model, based on restraints extracted by MTMDAT from limited proteolysis and modeled by HADDOCK, was compared to the published NMR structure, which relied on twelve unambiguous intermolecular NOE interactions. The MTMDAT-HADDOCK structure was of similar quality to structures generated using only chemical shift perturbation data derived by NMR titration experiments.
The new MTMDAT-HADDOCK pipeline enables direct high-throughput modeling of protein complexes from mass spectrometry data. MTMDAT-HADDOCK can be downloaded from http://www.ifm.liu.se/chemistry/molbiotech/maria_sunnerhagens_group/mtmdat/together with the manual and example files. The program is free for academic/non-commercial purposes.
期刊介绍:
BMC Structural Biology is an open access, peer-reviewed journal that considers articles on investigations into the structure of biological macromolecules, including solving structures, structural and functional analyses, and computational modeling.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
