Serafeim Alexopoulos , Anastasia Gkouskou , George Stravodimos , Anastasia S. Tsagkarakou , Ioannis Tsialtas , Demetres Katounis , Anna-Maria G. Psarra , Demetres Leonidas , Goutam Brahmachari , Joseph M. Hayes , Vasiliki Skamnaki
{"title":"香豆素类似物对糖原磷酸化酶激酶ATP结合位点的药理作用研究","authors":"Serafeim Alexopoulos , Anastasia Gkouskou , George Stravodimos , Anastasia S. Tsagkarakou , Ioannis Tsialtas , Demetres Katounis , Anna-Maria G. Psarra , Demetres Leonidas , Goutam Brahmachari , Joseph M. Hayes , Vasiliki Skamnaki","doi":"10.1016/j.crchbi.2022.100022","DOIUrl":null,"url":null,"abstract":"<div><p>Glycogen phosphorylase kinase (PhK) converts by phosphorylation, the inactive glycogen phosphorylase (GPb) into active GPa in the glycogenolytic pathway. It is a complex enzyme comprising of the catalytic (γ) and three regulatory subunits (α, β, δ) forming a hexadecamer with stoichiometry (αβγδ)<sub>4.</sub> Several studies have indicated PhK as a promising target for the development of antihyperglycemics as its inhibition blocks glycogenolysis in liver and a potential therapeutic target for cancer against pathological angiogenesis and tumor progression. The identification of compounds that inhibit the kinase through their direct binding to its catalytic site is an effective approach to identify bioactive molecules of therapeutic significance. Towards this, the structure of the N-terminal kinase domain (residues 1–298) of the catalytic γ subunit of PhK (PhKγtrnc) has been determined by X-ray crystallography while staurosporine and indirubin analogues have been characterized as potent inhibitors targeting the ATP binding site. In this study, a series of 38 synthetic analogues of naturally occurring coumarins were screened for inhibition of PhKγtrnc, <em>in vitro</em>, using a photometric assay. The <em>IC</em><sub>50</sub> values of the two most potent compounds were determined for PhKγtrnc and the pharmacologically relevant target, human liver isoform (PHKG2A). Their cellular efficacy and toxicity in HepG2 cells were further assessed <em>ex vivo</em>. Docking experiments and the structural comparison with previously described inhibitors reveal the binding mode of the coumarin scaffold at a no hinge region of the ATP site of PhK and the role of a conserved β3-Lys in binding. The experimental findings provide structural insights with implications to the kinase targeting and drug design.</p></div>","PeriodicalId":72747,"journal":{"name":"Current research in chemical biology","volume":"2 ","pages":"Article 100022"},"PeriodicalIF":0.0000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2666246922000040/pdfft?md5=aa4ec7dfb7558b10c5c83b49103718eb&pid=1-s2.0-S2666246922000040-main.pdf","citationCount":"2","resultStr":"{\"title\":\"The druggability of the ATP binding site of glycogen phosphorylase kinase probed by coumarin analogues\",\"authors\":\"Serafeim Alexopoulos , Anastasia Gkouskou , George Stravodimos , Anastasia S. Tsagkarakou , Ioannis Tsialtas , Demetres Katounis , Anna-Maria G. Psarra , Demetres Leonidas , Goutam Brahmachari , Joseph M. Hayes , Vasiliki Skamnaki\",\"doi\":\"10.1016/j.crchbi.2022.100022\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Glycogen phosphorylase kinase (PhK) converts by phosphorylation, the inactive glycogen phosphorylase (GPb) into active GPa in the glycogenolytic pathway. It is a complex enzyme comprising of the catalytic (γ) and three regulatory subunits (α, β, δ) forming a hexadecamer with stoichiometry (αβγδ)<sub>4.</sub> Several studies have indicated PhK as a promising target for the development of antihyperglycemics as its inhibition blocks glycogenolysis in liver and a potential therapeutic target for cancer against pathological angiogenesis and tumor progression. The identification of compounds that inhibit the kinase through their direct binding to its catalytic site is an effective approach to identify bioactive molecules of therapeutic significance. Towards this, the structure of the N-terminal kinase domain (residues 1–298) of the catalytic γ subunit of PhK (PhKγtrnc) has been determined by X-ray crystallography while staurosporine and indirubin analogues have been characterized as potent inhibitors targeting the ATP binding site. In this study, a series of 38 synthetic analogues of naturally occurring coumarins were screened for inhibition of PhKγtrnc, <em>in vitro</em>, using a photometric assay. The <em>IC</em><sub>50</sub> values of the two most potent compounds were determined for PhKγtrnc and the pharmacologically relevant target, human liver isoform (PHKG2A). Their cellular efficacy and toxicity in HepG2 cells were further assessed <em>ex vivo</em>. Docking experiments and the structural comparison with previously described inhibitors reveal the binding mode of the coumarin scaffold at a no hinge region of the ATP site of PhK and the role of a conserved β3-Lys in binding. The experimental findings provide structural insights with implications to the kinase targeting and drug design.</p></div>\",\"PeriodicalId\":72747,\"journal\":{\"name\":\"Current research in chemical biology\",\"volume\":\"2 \",\"pages\":\"Article 100022\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2022-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S2666246922000040/pdfft?md5=aa4ec7dfb7558b10c5c83b49103718eb&pid=1-s2.0-S2666246922000040-main.pdf\",\"citationCount\":\"2\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Current research in chemical biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2666246922000040\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/2/16 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current research in chemical biology","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2666246922000040","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/2/16 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
The druggability of the ATP binding site of glycogen phosphorylase kinase probed by coumarin analogues
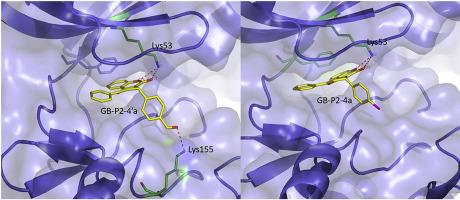
Glycogen phosphorylase kinase (PhK) converts by phosphorylation, the inactive glycogen phosphorylase (GPb) into active GPa in the glycogenolytic pathway. It is a complex enzyme comprising of the catalytic (γ) and three regulatory subunits (α, β, δ) forming a hexadecamer with stoichiometry (αβγδ)4. Several studies have indicated PhK as a promising target for the development of antihyperglycemics as its inhibition blocks glycogenolysis in liver and a potential therapeutic target for cancer against pathological angiogenesis and tumor progression. The identification of compounds that inhibit the kinase through their direct binding to its catalytic site is an effective approach to identify bioactive molecules of therapeutic significance. Towards this, the structure of the N-terminal kinase domain (residues 1–298) of the catalytic γ subunit of PhK (PhKγtrnc) has been determined by X-ray crystallography while staurosporine and indirubin analogues have been characterized as potent inhibitors targeting the ATP binding site. In this study, a series of 38 synthetic analogues of naturally occurring coumarins were screened for inhibition of PhKγtrnc, in vitro, using a photometric assay. The IC50 values of the two most potent compounds were determined for PhKγtrnc and the pharmacologically relevant target, human liver isoform (PHKG2A). Their cellular efficacy and toxicity in HepG2 cells were further assessed ex vivo. Docking experiments and the structural comparison with previously described inhibitors reveal the binding mode of the coumarin scaffold at a no hinge region of the ATP site of PhK and the role of a conserved β3-Lys in binding. The experimental findings provide structural insights with implications to the kinase targeting and drug design.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
