Orlando J Geli-Cruz, Carlos J Santos-Flores, Matias J Cafaro, Alex Ropelewski, Alex R Van Dam
{"title":"通过牛津纳米孔测序技术对无组装纳米孔读数映射器进行基准测试,以对复杂的千足虫肠道微生物群进行分类","authors":"Orlando J Geli-Cruz, Carlos J Santos-Flores, Matias J Cafaro, Alex Ropelewski, Alex R Van Dam","doi":"10.14440/jbm.2023.376","DOIUrl":null,"url":null,"abstract":"<p><p>Millipedes are key players in recycling leaf litter into soil in tropical ecosystems. To elucidate their gut microbiota, we collected millipedes from different municipalities of Puerto Rico. Here we aim to benchmark which method is best for metagenomic skimming of this highly complex millipede microbiome. We sequenced the gut DNA with Oxford Nanopore Technologies' (ONT) MinION sequencer, then analyzed the data using <i>MEGAN-LR</i>, <i>Kraken2</i> protein mode, <i>Kraken2</i> nucleotide mode, <i>GraphMap</i>, and <i>Minimap2</i> to classify these long ONT reads. From our two samples, we obtained a total of 87,110 and 99,749 ONT reads, respectively. <i>Kraken2</i> nucleotide mode classified the most reads compared to all other methods at the phylum and class taxonomic level, classifying 75% of the reads in the two samples, the other methods failed to assign enough reads to either phylum or class to yield asymptotes in the taxa rarefaction curves indicating that they required more sequencing depth to fully classify this community. The community is hyper diverse with all methods classifying 20-50 phyla in the two samples. There was significant overlap in the reads used and phyla classified between the five methods benchmarked. Our results suggest that <i>Kraken2</i> nucleotide mode is the most appropriate tool for the application of metagenomic skimming of this highly complex community.</p>","PeriodicalId":73618,"journal":{"name":"Journal of biological methods","volume":" ","pages":"e99010003"},"PeriodicalIF":0.0000,"publicationDate":"2023-08-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10627078/pdf/","citationCount":"0","resultStr":"{\"title\":\"Benchmarking assembly free nanopore read mappers to classify complex millipede gut microbiota via Oxford Nanopore Sequencing Technology.\",\"authors\":\"Orlando J Geli-Cruz, Carlos J Santos-Flores, Matias J Cafaro, Alex Ropelewski, Alex R Van Dam\",\"doi\":\"10.14440/jbm.2023.376\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Millipedes are key players in recycling leaf litter into soil in tropical ecosystems. To elucidate their gut microbiota, we collected millipedes from different municipalities of Puerto Rico. Here we aim to benchmark which method is best for metagenomic skimming of this highly complex millipede microbiome. We sequenced the gut DNA with Oxford Nanopore Technologies' (ONT) MinION sequencer, then analyzed the data using <i>MEGAN-LR</i>, <i>Kraken2</i> protein mode, <i>Kraken2</i> nucleotide mode, <i>GraphMap</i>, and <i>Minimap2</i> to classify these long ONT reads. From our two samples, we obtained a total of 87,110 and 99,749 ONT reads, respectively. <i>Kraken2</i> nucleotide mode classified the most reads compared to all other methods at the phylum and class taxonomic level, classifying 75% of the reads in the two samples, the other methods failed to assign enough reads to either phylum or class to yield asymptotes in the taxa rarefaction curves indicating that they required more sequencing depth to fully classify this community. The community is hyper diverse with all methods classifying 20-50 phyla in the two samples. There was significant overlap in the reads used and phyla classified between the five methods benchmarked. Our results suggest that <i>Kraken2</i> nucleotide mode is the most appropriate tool for the application of metagenomic skimming of this highly complex community.</p>\",\"PeriodicalId\":73618,\"journal\":{\"name\":\"Journal of biological methods\",\"volume\":\" \",\"pages\":\"e99010003\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-08-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10627078/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of biological methods\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.14440/jbm.2023.376\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of biological methods","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.14440/jbm.2023.376","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Benchmarking assembly free nanopore read mappers to classify complex millipede gut microbiota via Oxford Nanopore Sequencing Technology.
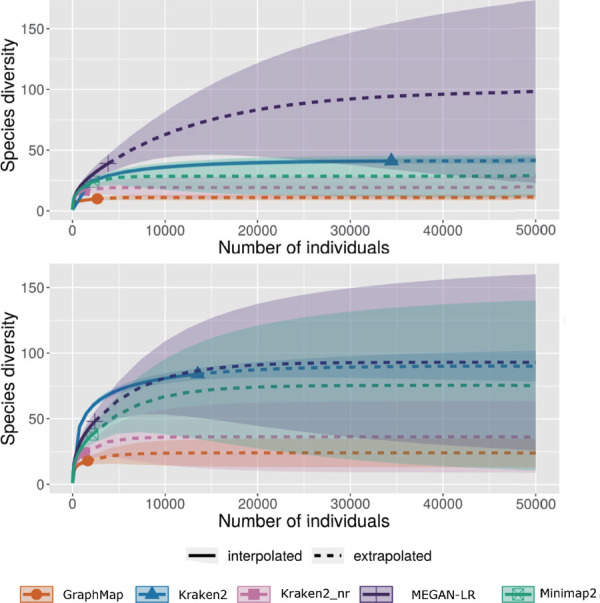
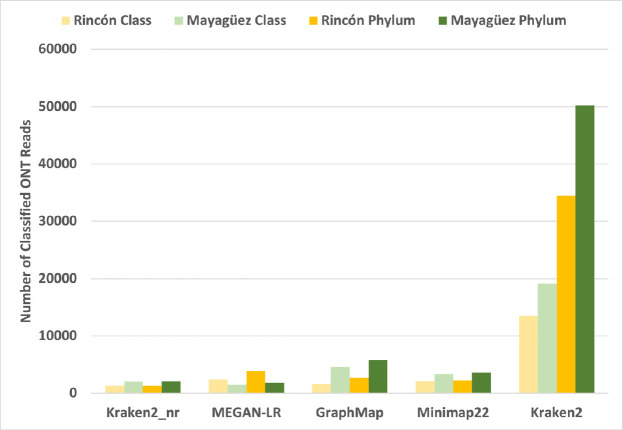
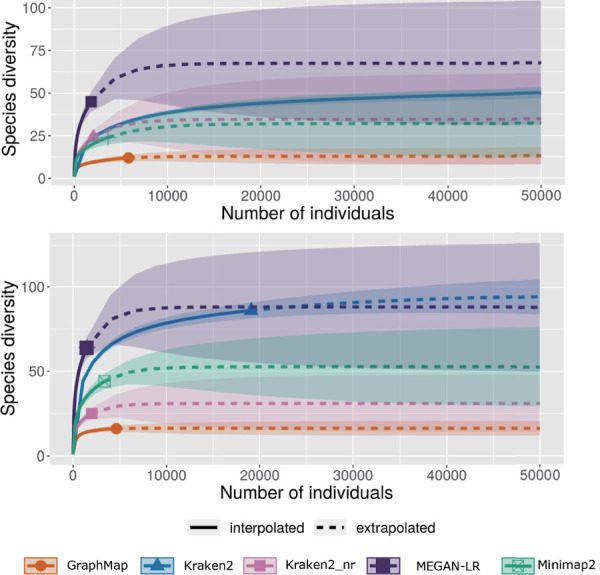
Millipedes are key players in recycling leaf litter into soil in tropical ecosystems. To elucidate their gut microbiota, we collected millipedes from different municipalities of Puerto Rico. Here we aim to benchmark which method is best for metagenomic skimming of this highly complex millipede microbiome. We sequenced the gut DNA with Oxford Nanopore Technologies' (ONT) MinION sequencer, then analyzed the data using MEGAN-LR, Kraken2 protein mode, Kraken2 nucleotide mode, GraphMap, and Minimap2 to classify these long ONT reads. From our two samples, we obtained a total of 87,110 and 99,749 ONT reads, respectively. Kraken2 nucleotide mode classified the most reads compared to all other methods at the phylum and class taxonomic level, classifying 75% of the reads in the two samples, the other methods failed to assign enough reads to either phylum or class to yield asymptotes in the taxa rarefaction curves indicating that they required more sequencing depth to fully classify this community. The community is hyper diverse with all methods classifying 20-50 phyla in the two samples. There was significant overlap in the reads used and phyla classified between the five methods benchmarked. Our results suggest that Kraken2 nucleotide mode is the most appropriate tool for the application of metagenomic skimming of this highly complex community.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
