{"title":"铁氧化还原蛋白样折叠起始片段信息的序列分析","authors":"Masanari Matsuoka, Takeshi Kikuchi","doi":"10.1186/1472-6807-14-15","DOIUrl":null,"url":null,"abstract":"<p>While some studies have shown that the 3D protein structures are more conservative than their amino acid sequences, other experimental studies have shown that even if two proteins share the same topology, they may have different folding pathways. There are many studies investigating this issue with molecular dynamics or Go-like model simulations, however, one should be able to obtain the same information by analyzing the proteins’ amino acid sequences, if the sequences contain all the information about the 3D structures. In this study, we use information about protein sequences to predict the location of their folding segments. We focus on proteins with a ferredoxin-like fold, which has a characteristic topology. Some of these proteins have different folding segments.</p><p>Despite the simplicity of our methods, we are able to correctly determine the experimentally identified folding segments by predicting the location of the compact regions considered to play an important role in structural formation. We also apply our sequence analyses to some homologues of each protein and confirm that there are highly conserved folding segments despite the homologues’ sequence diversity. These homologues have similar folding segments even though the homology of two proteins’ sequences is not so high.</p><p>Our analyses have proven useful for investigating the common or different folding features of the proteins studied.</p>","PeriodicalId":51240,"journal":{"name":"BMC Structural Biology","volume":"14 1","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2014-05-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1472-6807-14-15","citationCount":"14","resultStr":"{\"title\":\"Sequence analysis on the information of folding initiation segments in ferredoxin-like fold proteins\",\"authors\":\"Masanari Matsuoka, Takeshi Kikuchi\",\"doi\":\"10.1186/1472-6807-14-15\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>While some studies have shown that the 3D protein structures are more conservative than their amino acid sequences, other experimental studies have shown that even if two proteins share the same topology, they may have different folding pathways. There are many studies investigating this issue with molecular dynamics or Go-like model simulations, however, one should be able to obtain the same information by analyzing the proteins’ amino acid sequences, if the sequences contain all the information about the 3D structures. In this study, we use information about protein sequences to predict the location of their folding segments. We focus on proteins with a ferredoxin-like fold, which has a characteristic topology. Some of these proteins have different folding segments.</p><p>Despite the simplicity of our methods, we are able to correctly determine the experimentally identified folding segments by predicting the location of the compact regions considered to play an important role in structural formation. We also apply our sequence analyses to some homologues of each protein and confirm that there are highly conserved folding segments despite the homologues’ sequence diversity. These homologues have similar folding segments even though the homology of two proteins’ sequences is not so high.</p><p>Our analyses have proven useful for investigating the common or different folding features of the proteins studied.</p>\",\"PeriodicalId\":51240,\"journal\":{\"name\":\"BMC Structural Biology\",\"volume\":\"14 1\",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2014-05-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/1472-6807-14-15\",\"citationCount\":\"14\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Structural Biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://link.springer.com/article/10.1186/1472-6807-14-15\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Structural Biology","FirstCategoryId":"1085","ListUrlMain":"https://link.springer.com/article/10.1186/1472-6807-14-15","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
Sequence analysis on the information of folding initiation segments in ferredoxin-like fold proteins
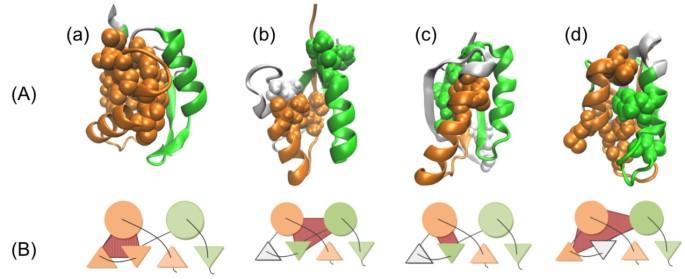
While some studies have shown that the 3D protein structures are more conservative than their amino acid sequences, other experimental studies have shown that even if two proteins share the same topology, they may have different folding pathways. There are many studies investigating this issue with molecular dynamics or Go-like model simulations, however, one should be able to obtain the same information by analyzing the proteins’ amino acid sequences, if the sequences contain all the information about the 3D structures. In this study, we use information about protein sequences to predict the location of their folding segments. We focus on proteins with a ferredoxin-like fold, which has a characteristic topology. Some of these proteins have different folding segments.
Despite the simplicity of our methods, we are able to correctly determine the experimentally identified folding segments by predicting the location of the compact regions considered to play an important role in structural formation. We also apply our sequence analyses to some homologues of each protein and confirm that there are highly conserved folding segments despite the homologues’ sequence diversity. These homologues have similar folding segments even though the homology of two proteins’ sequences is not so high.
Our analyses have proven useful for investigating the common or different folding features of the proteins studied.
期刊介绍:
BMC Structural Biology is an open access, peer-reviewed journal that considers articles on investigations into the structure of biological macromolecules, including solving structures, structural and functional analyses, and computational modeling.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
