{"title":"多参考计算如何改变催化化学?铜催化芳基碘化物和β-二酮偶联反应的DFT和CASPT2研究。","authors":"Nan He, Naoki Nakatani and Masahiko Hada","doi":"10.1039/D3CP03418F","DOIUrl":null,"url":null,"abstract":"<p >The molecular mechanism of a Cu-catalysed coupling reaction was theoretically studied using density functional theory (DFT) and the complete active space self-consistent field method followed by the second-order perturbation theory (CASSCF/CASPT2) to investigate the effects of the strong electron correlation of the Cu centre on the reaction profile. Both DFT and CASSCF/CASPT2 calculations showed that the catalytic cycle proceeds <em>via</em> an oxidative addition (OA) reaction, followed by a reductive elimination (RE) reaction, where OA is the rate-determining step. Although the DFT-calculated activation energies of the OA and RE steps are highly dependent on the choice of functionals, the CASSCF/CASPT2 results are less affected by the choice of DFT-optimised geometries. Therefore, with a careful assessment based on the CASSCF/CASPT2 single-point energy evaluation, an optimal choice of the DFT geometry is of good qualitative use for energetics at the CASPT2 level of theory. Based on the changes in the electron populations of the 3d orbitals during the OA and RE steps, the characteristic features of the DFT-calculated electronic structure were qualitatively consistent with those calculated using the CASSCF method. Further electronic structure analysis by the natural orbital occupancy of the CASSCF wavefunction showed that the ground state is almost single-reference in this system and the strong electron correlation effect of the Cu centre can be dealt with using the MP2 or CCSD method, too. However, the slightly smaller occupation numbers of the 3dπ orbital in the course of reactions suggested that the electron correlation effect of the Cu(<small>III</small>) centre appears through the interaction between the 3dπ orbital and the C–I antibonding σ* orbital in the OA step, and between the 3dπ orbital and the Cu–C antibonding σ* orbital in the RE step.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 42","pages":" 28871-28884"},"PeriodicalIF":2.9000,"publicationDate":"2023-10-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"How does multi-reference computation change the catalysis chemistry? DFT and CASPT2 studies of the Cu-catalysed coupling reactions between aryl iodides and β-diketones†\",\"authors\":\"Nan He, Naoki Nakatani and Masahiko Hada\",\"doi\":\"10.1039/D3CP03418F\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The molecular mechanism of a Cu-catalysed coupling reaction was theoretically studied using density functional theory (DFT) and the complete active space self-consistent field method followed by the second-order perturbation theory (CASSCF/CASPT2) to investigate the effects of the strong electron correlation of the Cu centre on the reaction profile. Both DFT and CASSCF/CASPT2 calculations showed that the catalytic cycle proceeds <em>via</em> an oxidative addition (OA) reaction, followed by a reductive elimination (RE) reaction, where OA is the rate-determining step. Although the DFT-calculated activation energies of the OA and RE steps are highly dependent on the choice of functionals, the CASSCF/CASPT2 results are less affected by the choice of DFT-optimised geometries. Therefore, with a careful assessment based on the CASSCF/CASPT2 single-point energy evaluation, an optimal choice of the DFT geometry is of good qualitative use for energetics at the CASPT2 level of theory. Based on the changes in the electron populations of the 3d orbitals during the OA and RE steps, the characteristic features of the DFT-calculated electronic structure were qualitatively consistent with those calculated using the CASSCF method. Further electronic structure analysis by the natural orbital occupancy of the CASSCF wavefunction showed that the ground state is almost single-reference in this system and the strong electron correlation effect of the Cu centre can be dealt with using the MP2 or CCSD method, too. However, the slightly smaller occupation numbers of the 3dπ orbital in the course of reactions suggested that the electron correlation effect of the Cu(<small>III</small>) centre appears through the interaction between the 3dπ orbital and the C–I antibonding σ* orbital in the OA step, and between the 3dπ orbital and the Cu–C antibonding σ* orbital in the RE step.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 42\",\"pages\":\" 28871-28884\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2023-10-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2023/cp/d3cp03418f\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2023/cp/d3cp03418f","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
How does multi-reference computation change the catalysis chemistry? DFT and CASPT2 studies of the Cu-catalysed coupling reactions between aryl iodides and β-diketones†
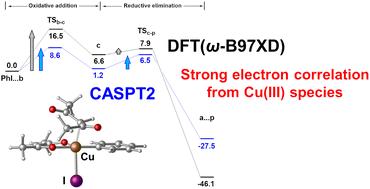
The molecular mechanism of a Cu-catalysed coupling reaction was theoretically studied using density functional theory (DFT) and the complete active space self-consistent field method followed by the second-order perturbation theory (CASSCF/CASPT2) to investigate the effects of the strong electron correlation of the Cu centre on the reaction profile. Both DFT and CASSCF/CASPT2 calculations showed that the catalytic cycle proceeds via an oxidative addition (OA) reaction, followed by a reductive elimination (RE) reaction, where OA is the rate-determining step. Although the DFT-calculated activation energies of the OA and RE steps are highly dependent on the choice of functionals, the CASSCF/CASPT2 results are less affected by the choice of DFT-optimised geometries. Therefore, with a careful assessment based on the CASSCF/CASPT2 single-point energy evaluation, an optimal choice of the DFT geometry is of good qualitative use for energetics at the CASPT2 level of theory. Based on the changes in the electron populations of the 3d orbitals during the OA and RE steps, the characteristic features of the DFT-calculated electronic structure were qualitatively consistent with those calculated using the CASSCF method. Further electronic structure analysis by the natural orbital occupancy of the CASSCF wavefunction showed that the ground state is almost single-reference in this system and the strong electron correlation effect of the Cu centre can be dealt with using the MP2 or CCSD method, too. However, the slightly smaller occupation numbers of the 3dπ orbital in the course of reactions suggested that the electron correlation effect of the Cu(III) centre appears through the interaction between the 3dπ orbital and the C–I antibonding σ* orbital in the OA step, and between the 3dπ orbital and the Cu–C antibonding σ* orbital in the RE step.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
