{"title":"罕见癌症型肾上腺皮质癌转化研究进展。","authors":"Chandrayee Ghosh, Jiangnan Hu, Electron Kebebew","doi":"10.1038/s41568-023-00623-0","DOIUrl":null,"url":null,"abstract":"Adrenocortical carcinoma is a rare malignancy with an annual worldwide incidence of 1–2 cases per 1 million and a 5-year survival rate of <60%. Although adrenocortical carcinoma is rare, such rare cancers account for approximately one third of patients diagnosed with cancer annually. In the past decade, there have been considerable advances in understanding the molecular basis of adrenocortical carcinoma. The genetic events associated with adrenocortical carcinoma in adults are distinct from those of paediatric cases, which are often associated with germline or somatic TP53 mutations and have a better prognosis. In adult primary adrenocortical carcinoma, the main somatic genetic alterations occur in genes that encode proteins involved in the WNT–β-catenin pathway, cell cycle and p53 apoptosis pathway, chromatin remodelling and telomere maintenance pathway, cAMP–protein kinase A (PKA) pathway or DNA transcription and RNA translation pathways. Recently, integrated molecular studies of adrenocortical carcinomas, which have characterized somatic mutations and the methylome as well as gene and microRNA expression profiles, have led to a molecular classification of these tumours that can predict prognosis and have helped to identify new therapeutic targets. In this Review, we summarize these recent translational research advances in adrenocortical carcinoma, which it is hoped could lead to improved patient diagnosis, treatment and outcome. Adrenocortical carcinoma is a rare endocrine cancer with a dismal survival rate and limited therapeutic options. This Review outlines the recent advances that have been made in the understanding of the molecular basis of adrenocortical carcinoma and what this means for the diagnosis and treatment of patients with this cancer type.","PeriodicalId":19055,"journal":{"name":"Nature Reviews Cancer","volume":"23 12","pages":"805-824"},"PeriodicalIF":66.8000,"publicationDate":"2023-10-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Advances in translational research of the rare cancer type adrenocortical carcinoma\",\"authors\":\"Chandrayee Ghosh, Jiangnan Hu, Electron Kebebew\",\"doi\":\"10.1038/s41568-023-00623-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Adrenocortical carcinoma is a rare malignancy with an annual worldwide incidence of 1–2 cases per 1 million and a 5-year survival rate of <60%. Although adrenocortical carcinoma is rare, such rare cancers account for approximately one third of patients diagnosed with cancer annually. In the past decade, there have been considerable advances in understanding the molecular basis of adrenocortical carcinoma. The genetic events associated with adrenocortical carcinoma in adults are distinct from those of paediatric cases, which are often associated with germline or somatic TP53 mutations and have a better prognosis. In adult primary adrenocortical carcinoma, the main somatic genetic alterations occur in genes that encode proteins involved in the WNT–β-catenin pathway, cell cycle and p53 apoptosis pathway, chromatin remodelling and telomere maintenance pathway, cAMP–protein kinase A (PKA) pathway or DNA transcription and RNA translation pathways. Recently, integrated molecular studies of adrenocortical carcinomas, which have characterized somatic mutations and the methylome as well as gene and microRNA expression profiles, have led to a molecular classification of these tumours that can predict prognosis and have helped to identify new therapeutic targets. In this Review, we summarize these recent translational research advances in adrenocortical carcinoma, which it is hoped could lead to improved patient diagnosis, treatment and outcome. Adrenocortical carcinoma is a rare endocrine cancer with a dismal survival rate and limited therapeutic options. This Review outlines the recent advances that have been made in the understanding of the molecular basis of adrenocortical carcinoma and what this means for the diagnosis and treatment of patients with this cancer type.\",\"PeriodicalId\":19055,\"journal\":{\"name\":\"Nature Reviews Cancer\",\"volume\":\"23 12\",\"pages\":\"805-824\"},\"PeriodicalIF\":66.8000,\"publicationDate\":\"2023-10-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature Reviews Cancer\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.nature.com/articles/s41568-023-00623-0\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"ONCOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Reviews Cancer","FirstCategoryId":"3","ListUrlMain":"https://www.nature.com/articles/s41568-023-00623-0","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ONCOLOGY","Score":null,"Total":0}
Advances in translational research of the rare cancer type adrenocortical carcinoma
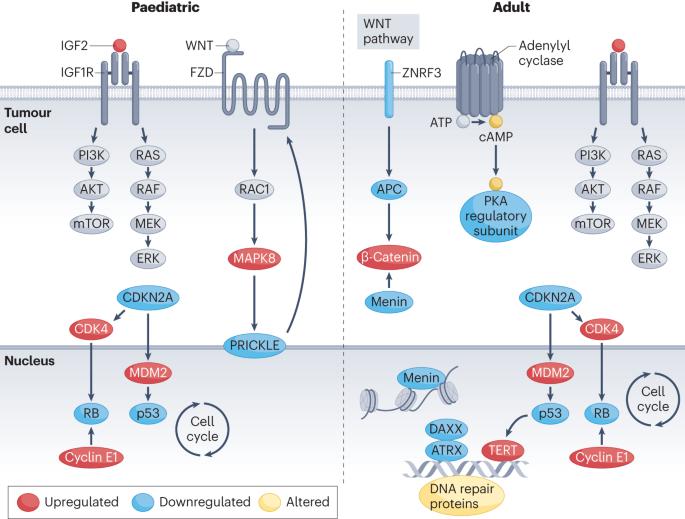
Adrenocortical carcinoma is a rare malignancy with an annual worldwide incidence of 1–2 cases per 1 million and a 5-year survival rate of <60%. Although adrenocortical carcinoma is rare, such rare cancers account for approximately one third of patients diagnosed with cancer annually. In the past decade, there have been considerable advances in understanding the molecular basis of adrenocortical carcinoma. The genetic events associated with adrenocortical carcinoma in adults are distinct from those of paediatric cases, which are often associated with germline or somatic TP53 mutations and have a better prognosis. In adult primary adrenocortical carcinoma, the main somatic genetic alterations occur in genes that encode proteins involved in the WNT–β-catenin pathway, cell cycle and p53 apoptosis pathway, chromatin remodelling and telomere maintenance pathway, cAMP–protein kinase A (PKA) pathway or DNA transcription and RNA translation pathways. Recently, integrated molecular studies of adrenocortical carcinomas, which have characterized somatic mutations and the methylome as well as gene and microRNA expression profiles, have led to a molecular classification of these tumours that can predict prognosis and have helped to identify new therapeutic targets. In this Review, we summarize these recent translational research advances in adrenocortical carcinoma, which it is hoped could lead to improved patient diagnosis, treatment and outcome. Adrenocortical carcinoma is a rare endocrine cancer with a dismal survival rate and limited therapeutic options. This Review outlines the recent advances that have been made in the understanding of the molecular basis of adrenocortical carcinoma and what this means for the diagnosis and treatment of patients with this cancer type.
期刊介绍:
Nature Reviews Cancer, a part of the Nature Reviews portfolio of journals, aims to be the premier source of reviews and commentaries for the scientific communities it serves. The correct abbreviation for abstracting and indexing purposes is Nat. Rev. Cancer. The international standard serial numbers (ISSN) for Nature Reviews Cancer are 1474-175X (print) and 1474-1768 (online). Unlike other journals, Nature Reviews Cancer does not have an external editorial board. Instead, all editorial decisions are made by a team of full-time professional editors who are PhD-level scientists. The journal publishes Research Highlights, Comments, Reviews, and Perspectives relevant to cancer researchers, ensuring that the articles reach the widest possible audience due to their broad scope.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
