{"title":"用微溶剂化方法模拟VCD光谱分析中的溶质-溶剂相互作用。","authors":"Christian Merten","doi":"10.1039/D3CP03408A","DOIUrl":null,"url":null,"abstract":"<p >Vibrational circular dichroism (VCD) spectroscopy has become an important part of the (stereo-)chemists’ toolbox as a reliable method for the determination of absolute configurations. Being the chiroptical version of infrared spectroscopy, it has also been recognized as being very sensitive to conformational changes and intermolecular interactions. This sensitivity originates from the fact that the VCD spectra of individual conformers are often more different than their IR spectra, so that changes in conformational distributions or band positions and intensities become more pronounced. What is an advantage for studies focussing on intermolecular interactions can, however, quickly turn into a major obstacle during AC determinations: solute–solvent interactions can have a strong influence on spectral signatures and they must be accurately treated when simulating VCD and IR spectra. In this perspective, we showcase selected examples which exhibit particularly pronounced solvent effects. It is demonstrated that it is typically sufficient to model solute–solvent interactions by placing single solvent molecules near hydrogen bonding sites of the solute and subsequently use the optimized structures for spectra simulations. This micro-solvation approach works reasonably well for medium-sized, not too conformationally flexible molecules. We thus also discuss its limitations and outline the next steps that method development needs to take in order to further improve the workflows for VCD spectra predictions.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 43","pages":" 29404-29414"},"PeriodicalIF":2.9000,"publicationDate":"2023-10-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Modelling solute–solvent interactions in VCD spectra analysis with the micro-solvation approach\",\"authors\":\"Christian Merten\",\"doi\":\"10.1039/D3CP03408A\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Vibrational circular dichroism (VCD) spectroscopy has become an important part of the (stereo-)chemists’ toolbox as a reliable method for the determination of absolute configurations. Being the chiroptical version of infrared spectroscopy, it has also been recognized as being very sensitive to conformational changes and intermolecular interactions. This sensitivity originates from the fact that the VCD spectra of individual conformers are often more different than their IR spectra, so that changes in conformational distributions or band positions and intensities become more pronounced. What is an advantage for studies focussing on intermolecular interactions can, however, quickly turn into a major obstacle during AC determinations: solute–solvent interactions can have a strong influence on spectral signatures and they must be accurately treated when simulating VCD and IR spectra. In this perspective, we showcase selected examples which exhibit particularly pronounced solvent effects. It is demonstrated that it is typically sufficient to model solute–solvent interactions by placing single solvent molecules near hydrogen bonding sites of the solute and subsequently use the optimized structures for spectra simulations. This micro-solvation approach works reasonably well for medium-sized, not too conformationally flexible molecules. We thus also discuss its limitations and outline the next steps that method development needs to take in order to further improve the workflows for VCD spectra predictions.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 43\",\"pages\":\" 29404-29414\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2023-10-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2023/cp/d3cp03408a\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2023/cp/d3cp03408a","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Modelling solute–solvent interactions in VCD spectra analysis with the micro-solvation approach
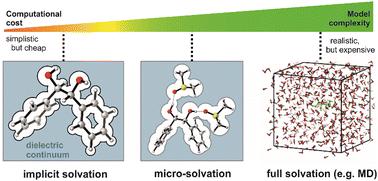
Vibrational circular dichroism (VCD) spectroscopy has become an important part of the (stereo-)chemists’ toolbox as a reliable method for the determination of absolute configurations. Being the chiroptical version of infrared spectroscopy, it has also been recognized as being very sensitive to conformational changes and intermolecular interactions. This sensitivity originates from the fact that the VCD spectra of individual conformers are often more different than their IR spectra, so that changes in conformational distributions or band positions and intensities become more pronounced. What is an advantage for studies focussing on intermolecular interactions can, however, quickly turn into a major obstacle during AC determinations: solute–solvent interactions can have a strong influence on spectral signatures and they must be accurately treated when simulating VCD and IR spectra. In this perspective, we showcase selected examples which exhibit particularly pronounced solvent effects. It is demonstrated that it is typically sufficient to model solute–solvent interactions by placing single solvent molecules near hydrogen bonding sites of the solute and subsequently use the optimized structures for spectra simulations. This micro-solvation approach works reasonably well for medium-sized, not too conformationally flexible molecules. We thus also discuss its limitations and outline the next steps that method development needs to take in order to further improve the workflows for VCD spectra predictions.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
