Katherine D Arias, Juan Pablo Gutiérrez, Iván Fernández, Isabel Álvarez, Félix Goyache
{"title":"Gochu Asturcelta猪一个小家系的自接合性研究。","authors":"Katherine D Arias, Juan Pablo Gutiérrez, Iván Fernández, Isabel Álvarez, Félix Goyache","doi":"10.1186/s12711-023-00846-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>In spite of the availability of single nucleotide polymorphism (SNP) array data, differentiation between observed homozygosity and that caused by mating between relatives (autozygosity) introduces major difficulties. Homozygosity estimators show large variation due to different causes, namely, Mendelian sampling, population structure, and differences among chromosomes. Therefore, the ascertainment of how inbreeding is reflected in the genome is still an issue. The aim of this research was to study the usefulness of genomic information for the assessment of genetic diversity in the highly endangered Gochu Asturcelta pig breed. Pedigree depth varied from 0 (founders) to 4 equivalent discrete generations (t). Four homozygosity parameters (runs of homozygosity, F<sub>ROH</sub>; heterozygosity-rich regions, F<sub>HRR</sub>; Li and Horvitz's, F<sub>LH</sub>; and Yang and colleague's F<sub>YAN</sub>) were computed for each individual, adjusted for the variability in the base population (BP; six individuals) and further jackknifed over autosomes. Individual increases in homozygosity (depending on t) and increases in pairwise homozygosity (i.e., increase in the parents' mean) were computed for each individual in the pedigree, and effective population size (N<sub>e</sub>) was computed for five subpopulations (cohorts). Genealogical parameters (individual inbreeding, individual increase in inbreeding, and N<sub>e</sub>) were used for comparisons.</p><p><strong>Results: </strong>The mean F was 0.120 ± 0.074 and the mean BP-adjusted homozygosity ranged from 0.099 ± 0.081 (F<sub>LH</sub>) to 0.152 ± 0.075 (F<sub>YAN</sub>). After jackknifing, the mean values were slightly lower. The increase in pairwise homozygosity tended to be twofold higher than the corresponding individual increase in homozygosity values. When compared with genealogical estimates, estimates of N<sub>e</sub> obtained using F<sub>YAN</sub> tended to have low root-mean-squared errors. However, N<sub>e</sub> estimates based on increases in pairwise homozygosity using both F<sub>ROH</sub> and F<sub>HRR</sub> estimates of genomic inbreeding had lower root-mean-squared errors.</p><p><strong>Conclusions: </strong>Parameters characterizing homozygosity may not accurately depict losses of variability in small populations in which breeding policy prohibits matings between close relatives. After BP adjustment, the performance of F<sub>ROH</sub> and F<sub>HRR</sub> was highly consistent. Assuming that an increase in homozygosity depends only on pedigree depth can lead to underestimating it in populations with shallow pedigrees. An increase in pairwise homozygosity computed from either F<sub>ROH</sub> or F<sub>HRR</sub> is a promising approach for characterizing autozygosity.</p>","PeriodicalId":55120,"journal":{"name":"Genetics Selection Evolution","volume":"55 1","pages":"74"},"PeriodicalIF":3.1000,"publicationDate":"2023-10-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10601182/pdf/","citationCount":"0","resultStr":"{\"title\":\"Approaching autozygosity in a small pedigree of Gochu Asturcelta pigs.\",\"authors\":\"Katherine D Arias, Juan Pablo Gutiérrez, Iván Fernández, Isabel Álvarez, Félix Goyache\",\"doi\":\"10.1186/s12711-023-00846-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>In spite of the availability of single nucleotide polymorphism (SNP) array data, differentiation between observed homozygosity and that caused by mating between relatives (autozygosity) introduces major difficulties. Homozygosity estimators show large variation due to different causes, namely, Mendelian sampling, population structure, and differences among chromosomes. Therefore, the ascertainment of how inbreeding is reflected in the genome is still an issue. The aim of this research was to study the usefulness of genomic information for the assessment of genetic diversity in the highly endangered Gochu Asturcelta pig breed. Pedigree depth varied from 0 (founders) to 4 equivalent discrete generations (t). Four homozygosity parameters (runs of homozygosity, F<sub>ROH</sub>; heterozygosity-rich regions, F<sub>HRR</sub>; Li and Horvitz's, F<sub>LH</sub>; and Yang and colleague's F<sub>YAN</sub>) were computed for each individual, adjusted for the variability in the base population (BP; six individuals) and further jackknifed over autosomes. Individual increases in homozygosity (depending on t) and increases in pairwise homozygosity (i.e., increase in the parents' mean) were computed for each individual in the pedigree, and effective population size (N<sub>e</sub>) was computed for five subpopulations (cohorts). Genealogical parameters (individual inbreeding, individual increase in inbreeding, and N<sub>e</sub>) were used for comparisons.</p><p><strong>Results: </strong>The mean F was 0.120 ± 0.074 and the mean BP-adjusted homozygosity ranged from 0.099 ± 0.081 (F<sub>LH</sub>) to 0.152 ± 0.075 (F<sub>YAN</sub>). After jackknifing, the mean values were slightly lower. The increase in pairwise homozygosity tended to be twofold higher than the corresponding individual increase in homozygosity values. When compared with genealogical estimates, estimates of N<sub>e</sub> obtained using F<sub>YAN</sub> tended to have low root-mean-squared errors. However, N<sub>e</sub> estimates based on increases in pairwise homozygosity using both F<sub>ROH</sub> and F<sub>HRR</sub> estimates of genomic inbreeding had lower root-mean-squared errors.</p><p><strong>Conclusions: </strong>Parameters characterizing homozygosity may not accurately depict losses of variability in small populations in which breeding policy prohibits matings between close relatives. After BP adjustment, the performance of F<sub>ROH</sub> and F<sub>HRR</sub> was highly consistent. Assuming that an increase in homozygosity depends only on pedigree depth can lead to underestimating it in populations with shallow pedigrees. An increase in pairwise homozygosity computed from either F<sub>ROH</sub> or F<sub>HRR</sub> is a promising approach for characterizing autozygosity.</p>\",\"PeriodicalId\":55120,\"journal\":{\"name\":\"Genetics Selection Evolution\",\"volume\":\"55 1\",\"pages\":\"74\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2023-10-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10601182/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genetics Selection Evolution\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s12711-023-00846-7\",\"RegionNum\":1,\"RegionCategory\":\"农林科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"AGRICULTURE, DAIRY & ANIMAL SCIENCE\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genetics Selection Evolution","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12711-023-00846-7","RegionNum":1,"RegionCategory":"农林科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"AGRICULTURE, DAIRY & ANIMAL SCIENCE","Score":null,"Total":0}
Approaching autozygosity in a small pedigree of Gochu Asturcelta pigs.
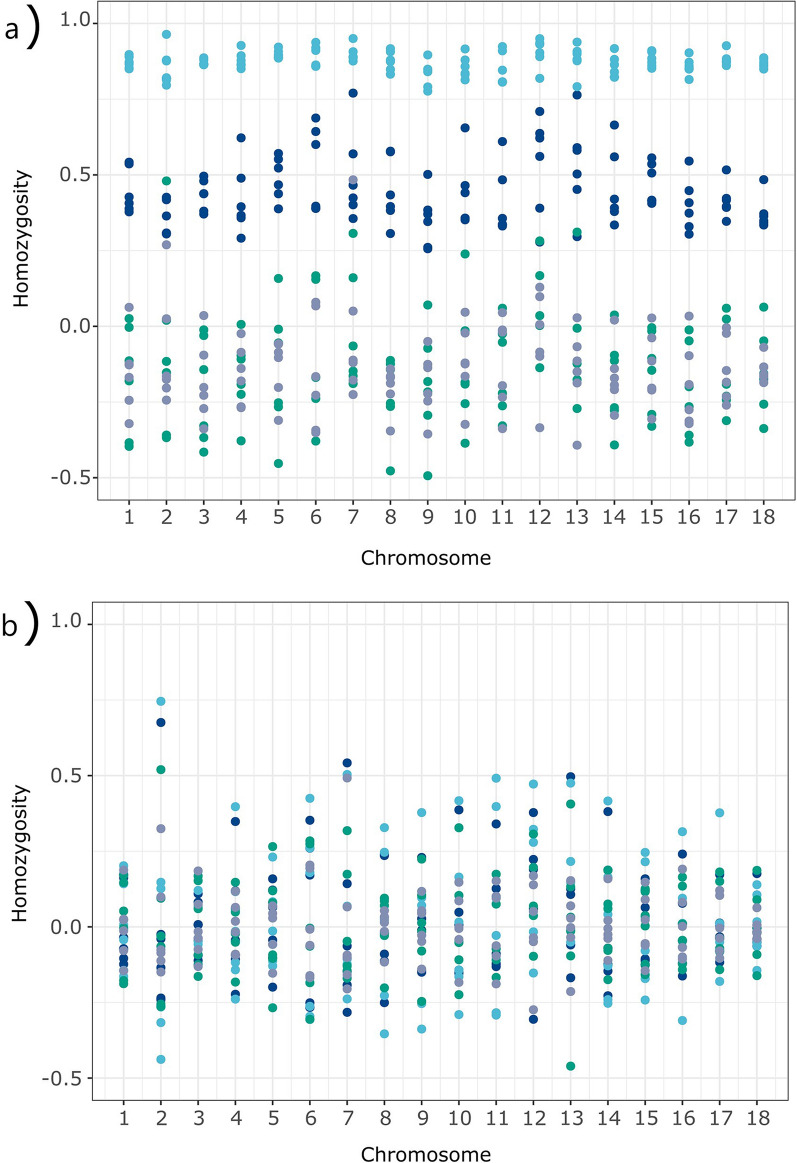
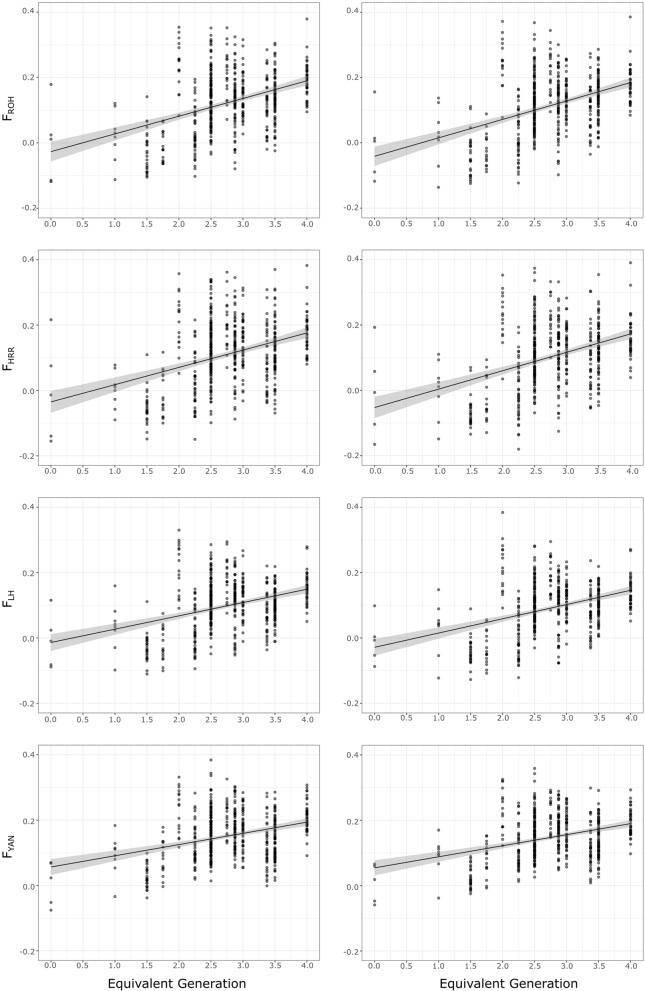
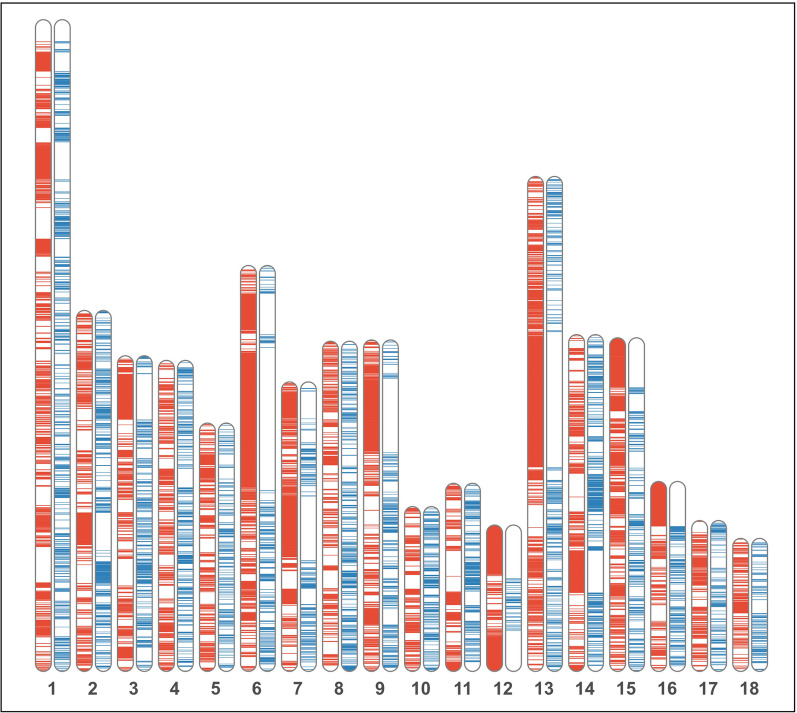
Background: In spite of the availability of single nucleotide polymorphism (SNP) array data, differentiation between observed homozygosity and that caused by mating between relatives (autozygosity) introduces major difficulties. Homozygosity estimators show large variation due to different causes, namely, Mendelian sampling, population structure, and differences among chromosomes. Therefore, the ascertainment of how inbreeding is reflected in the genome is still an issue. The aim of this research was to study the usefulness of genomic information for the assessment of genetic diversity in the highly endangered Gochu Asturcelta pig breed. Pedigree depth varied from 0 (founders) to 4 equivalent discrete generations (t). Four homozygosity parameters (runs of homozygosity, FROH; heterozygosity-rich regions, FHRR; Li and Horvitz's, FLH; and Yang and colleague's FYAN) were computed for each individual, adjusted for the variability in the base population (BP; six individuals) and further jackknifed over autosomes. Individual increases in homozygosity (depending on t) and increases in pairwise homozygosity (i.e., increase in the parents' mean) were computed for each individual in the pedigree, and effective population size (Ne) was computed for five subpopulations (cohorts). Genealogical parameters (individual inbreeding, individual increase in inbreeding, and Ne) were used for comparisons.
Results: The mean F was 0.120 ± 0.074 and the mean BP-adjusted homozygosity ranged from 0.099 ± 0.081 (FLH) to 0.152 ± 0.075 (FYAN). After jackknifing, the mean values were slightly lower. The increase in pairwise homozygosity tended to be twofold higher than the corresponding individual increase in homozygosity values. When compared with genealogical estimates, estimates of Ne obtained using FYAN tended to have low root-mean-squared errors. However, Ne estimates based on increases in pairwise homozygosity using both FROH and FHRR estimates of genomic inbreeding had lower root-mean-squared errors.
Conclusions: Parameters characterizing homozygosity may not accurately depict losses of variability in small populations in which breeding policy prohibits matings between close relatives. After BP adjustment, the performance of FROH and FHRR was highly consistent. Assuming that an increase in homozygosity depends only on pedigree depth can lead to underestimating it in populations with shallow pedigrees. An increase in pairwise homozygosity computed from either FROH or FHRR is a promising approach for characterizing autozygosity.
期刊介绍:
Genetics Selection Evolution invites basic, applied and methodological content that will aid the current understanding and the utilization of genetic variability in domestic animal species. Although the focus is on domestic animal species, research on other species is invited if it contributes to the understanding of the use of genetic variability in domestic animals. Genetics Selection Evolution publishes results from all levels of study, from the gene to the quantitative trait, from the individual to the population, the breed or the species. Contributions concerning both the biological approach, from molecular genetics to quantitative genetics, as well as the mathematical approach, from population genetics to statistics, are welcome. Specific areas of interest include but are not limited to: gene and QTL identification, mapping and characterization, analysis of new phenotypes, high-throughput SNP data analysis, functional genomics, cytogenetics, genetic diversity of populations and breeds, genetic evaluation, applied and experimental selection, genomic selection, selection efficiency, and statistical methodology for the genetic analysis of phenotypes with quantitative and mixed inheritance.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
