{"title":"MULTICOM蛋白结构预测系统的设计与对标","authors":"Jilong Li, Xin Deng, Jesse Eickholt, Jianlin Cheng","doi":"10.1186/1472-6807-13-2","DOIUrl":null,"url":null,"abstract":"<p>Predicting protein structure from sequence is one of the most significant and challenging problems in bioinformatics. Numerous bioinformatics techniques and tools have been developed to tackle almost every aspect of protein structure prediction ranging from structural feature prediction, template identification and query-template alignment to structure sampling, model quality assessment, and model refinement. How to synergistically select, integrate and improve the strengths of the complementary techniques at each prediction stage and build a high-performance system is becoming a critical issue for constructing a successful, competitive protein structure predictor.</p><p>Over the past several years, we have constructed a standalone protein structure prediction system MULTICOM that combines multiple sources of information and complementary methods at all five stages of the protein structure prediction process including template identification, template combination, model generation, model assessment, and model refinement. The system was blindly tested during the ninth Critical Assessment of Techniques for Protein Structure Prediction (CASP9) in 2010 and yielded very good performance. In addition to studying the overall performance on the CASP9 benchmark, we thoroughly investigated the performance and contributions of each component at each stage of prediction.</p><p>Our comprehensive and comparative study not only provides useful and practical insights about how to select, improve, and integrate complementary methods to build a cutting-edge protein structure prediction system but also identifies a few new sources of information that may help improve the design of a protein structure prediction system. Several components used in the MULTICOM system are available at: http://sysbio.rnet.missouri.edu/multicom_toolbox/.</p>","PeriodicalId":51240,"journal":{"name":"BMC Structural Biology","volume":"13 1","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2013-02-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1472-6807-13-2","citationCount":"29","resultStr":"{\"title\":\"Designing and benchmarking the MULTICOM protein structure prediction system\",\"authors\":\"Jilong Li, Xin Deng, Jesse Eickholt, Jianlin Cheng\",\"doi\":\"10.1186/1472-6807-13-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Predicting protein structure from sequence is one of the most significant and challenging problems in bioinformatics. Numerous bioinformatics techniques and tools have been developed to tackle almost every aspect of protein structure prediction ranging from structural feature prediction, template identification and query-template alignment to structure sampling, model quality assessment, and model refinement. How to synergistically select, integrate and improve the strengths of the complementary techniques at each prediction stage and build a high-performance system is becoming a critical issue for constructing a successful, competitive protein structure predictor.</p><p>Over the past several years, we have constructed a standalone protein structure prediction system MULTICOM that combines multiple sources of information and complementary methods at all five stages of the protein structure prediction process including template identification, template combination, model generation, model assessment, and model refinement. The system was blindly tested during the ninth Critical Assessment of Techniques for Protein Structure Prediction (CASP9) in 2010 and yielded very good performance. In addition to studying the overall performance on the CASP9 benchmark, we thoroughly investigated the performance and contributions of each component at each stage of prediction.</p><p>Our comprehensive and comparative study not only provides useful and practical insights about how to select, improve, and integrate complementary methods to build a cutting-edge protein structure prediction system but also identifies a few new sources of information that may help improve the design of a protein structure prediction system. Several components used in the MULTICOM system are available at: http://sysbio.rnet.missouri.edu/multicom_toolbox/.</p>\",\"PeriodicalId\":51240,\"journal\":{\"name\":\"BMC Structural Biology\",\"volume\":\"13 1\",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2013-02-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/1472-6807-13-2\",\"citationCount\":\"29\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Structural Biology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://link.springer.com/article/10.1186/1472-6807-13-2\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"Biochemistry, Genetics and Molecular Biology\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Structural Biology","FirstCategoryId":"1085","ListUrlMain":"https://link.springer.com/article/10.1186/1472-6807-13-2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 29
摘要
从序列中预测蛋白质结构是生物信息学中最重要和最具挑战性的问题之一。已经开发了许多生物信息学技术和工具来解决蛋白质结构预测的几乎每个方面,从结构特征预测,模板识别和查询模板比对到结构采样,模型质量评估和模型改进。如何在各个预测阶段协同选择、整合和提高互补技术的优势,构建一个高性能的系统,成为构建一个成功的、有竞争力的蛋白质结构预测器的关键问题。在过去的几年里,我们构建了一个独立的蛋白质结构预测系统MULTICOM,它结合了多种信息来源和互补的方法,在蛋白质结构预测过程的所有五个阶段,包括模板识别、模板组合、模型生成、模型评估和模型优化。该系统在2010年第九届CASP9蛋白结构预测技术关键评估(Critical Assessment of Techniques for Protein Structure Prediction, CASP9)中进行了盲测,取得了很好的效果。除了研究CASP9基准上的整体性能外,我们还深入研究了每个组件在预测的每个阶段的性能和贡献。我们的综合比较研究不仅为如何选择、改进和整合互补方法来构建前沿的蛋白质结构预测系统提供了有用和实用的见解,而且还确定了一些新的信息来源,可能有助于改进蛋白质结构预测系统的设计。MULTICOM系统中使用的几个组件可在:http://sysbio.rnet.missouri.edu/multicom_toolbox/上获得。
Designing and benchmarking the MULTICOM protein structure prediction system
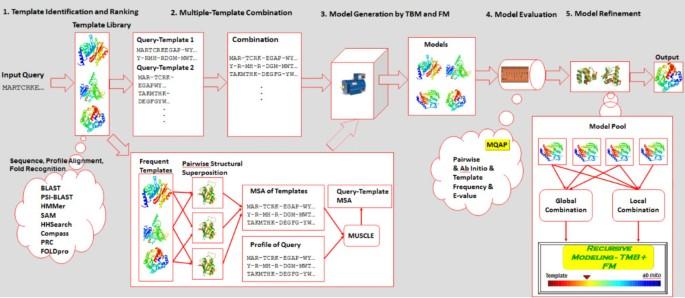
Predicting protein structure from sequence is one of the most significant and challenging problems in bioinformatics. Numerous bioinformatics techniques and tools have been developed to tackle almost every aspect of protein structure prediction ranging from structural feature prediction, template identification and query-template alignment to structure sampling, model quality assessment, and model refinement. How to synergistically select, integrate and improve the strengths of the complementary techniques at each prediction stage and build a high-performance system is becoming a critical issue for constructing a successful, competitive protein structure predictor.
Over the past several years, we have constructed a standalone protein structure prediction system MULTICOM that combines multiple sources of information and complementary methods at all five stages of the protein structure prediction process including template identification, template combination, model generation, model assessment, and model refinement. The system was blindly tested during the ninth Critical Assessment of Techniques for Protein Structure Prediction (CASP9) in 2010 and yielded very good performance. In addition to studying the overall performance on the CASP9 benchmark, we thoroughly investigated the performance and contributions of each component at each stage of prediction.
Our comprehensive and comparative study not only provides useful and practical insights about how to select, improve, and integrate complementary methods to build a cutting-edge protein structure prediction system but also identifies a few new sources of information that may help improve the design of a protein structure prediction system. Several components used in the MULTICOM system are available at: http://sysbio.rnet.missouri.edu/multicom_toolbox/.
期刊介绍:
BMC Structural Biology is an open access, peer-reviewed journal that considers articles on investigations into the structure of biological macromolecules, including solving structures, structural and functional analyses, and computational modeling.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
