{"title":"定向NMR光谱的模拟:结合分子动力学和化学位移张量计算。","authors":"Ulrich Sternberg, Raiker Witter","doi":"10.1002/mrc.5403","DOIUrl":null,"url":null,"abstract":"<p>Solid state NMR is widely used to study the orientation and other structural features of proteins and peptides in lipid bilayers. Using data obtained by PISEMA (Polarization Inversion Spin Exchange at Magic Angle) experiments, periodic spectral patterns arise from well-aligned α-helical molecules. Significant problems in the interpretation of PISEMA spectra may arise for systems that do not form perfectly defined secondary structures, like α-helices, or the signal pattern is disturbed by molecular motion. Here, we present a new method that combines molecular dynamics simulation with tensorial orientational constraints (MDOC) and chemical shift tensor calculations for the simulation and interpretation of PISEMA-like spectra. The calculations include the spectra arising from non α-helical molecules and molecules with non-uniform intrinsic mobility. In a first step, dipolar or quadrupolar interaction tensors drive molecular rotations and reorientations to obtain the proper mean values as observed in corresponding NMR experiments. In a second step, the coordinate snapshots of the MDOC simulations are geometry optimized with the isotropic <sup>15</sup>N chemical shifts as constraints using Bond Polarization Theory (BPT) to provide reliable <sup>15</sup>N CS tensor data. The averaged dipolar <sup>1</sup>H-<sup>15</sup>N couplings and the <i>δ</i><sub><i>zz</i></sub> tensor components can then be combined to simulate PISEMA patterns. We apply this method to the ß-helical peptide gramicidin A (gA) and demonstrate that this method enables the assignment of most PISEMA resonances. In addition, MDOC simulations provide local order parameters for the calculated sites. These local order parameters reveal large differences in backbone mobility between L- and D-amino acids of gA.</p>","PeriodicalId":18142,"journal":{"name":"Magnetic Resonance in Chemistry","volume":"62 3","pages":"125-144"},"PeriodicalIF":1.4000,"publicationDate":"2023-10-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/mrc.5403","citationCount":"0","resultStr":"{\"title\":\"Simulation of oriented NMR spectra: Combining molecular dynamics and chemical shift tensor calculations\",\"authors\":\"Ulrich Sternberg, Raiker Witter\",\"doi\":\"10.1002/mrc.5403\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Solid state NMR is widely used to study the orientation and other structural features of proteins and peptides in lipid bilayers. Using data obtained by PISEMA (Polarization Inversion Spin Exchange at Magic Angle) experiments, periodic spectral patterns arise from well-aligned α-helical molecules. Significant problems in the interpretation of PISEMA spectra may arise for systems that do not form perfectly defined secondary structures, like α-helices, or the signal pattern is disturbed by molecular motion. Here, we present a new method that combines molecular dynamics simulation with tensorial orientational constraints (MDOC) and chemical shift tensor calculations for the simulation and interpretation of PISEMA-like spectra. The calculations include the spectra arising from non α-helical molecules and molecules with non-uniform intrinsic mobility. In a first step, dipolar or quadrupolar interaction tensors drive molecular rotations and reorientations to obtain the proper mean values as observed in corresponding NMR experiments. In a second step, the coordinate snapshots of the MDOC simulations are geometry optimized with the isotropic <sup>15</sup>N chemical shifts as constraints using Bond Polarization Theory (BPT) to provide reliable <sup>15</sup>N CS tensor data. The averaged dipolar <sup>1</sup>H-<sup>15</sup>N couplings and the <i>δ</i><sub><i>zz</i></sub> tensor components can then be combined to simulate PISEMA patterns. We apply this method to the ß-helical peptide gramicidin A (gA) and demonstrate that this method enables the assignment of most PISEMA resonances. In addition, MDOC simulations provide local order parameters for the calculated sites. These local order parameters reveal large differences in backbone mobility between L- and D-amino acids of gA.</p>\",\"PeriodicalId\":18142,\"journal\":{\"name\":\"Magnetic Resonance in Chemistry\",\"volume\":\"62 3\",\"pages\":\"125-144\"},\"PeriodicalIF\":1.4000,\"publicationDate\":\"2023-10-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/mrc.5403\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Magnetic Resonance in Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://analyticalsciencejournals.onlinelibrary.wiley.com/doi/10.1002/mrc.5403\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Magnetic Resonance in Chemistry","FirstCategoryId":"92","ListUrlMain":"https://analyticalsciencejournals.onlinelibrary.wiley.com/doi/10.1002/mrc.5403","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Simulation of oriented NMR spectra: Combining molecular dynamics and chemical shift tensor calculations
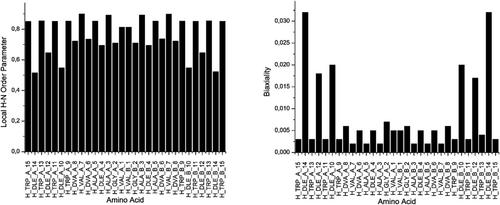
Solid state NMR is widely used to study the orientation and other structural features of proteins and peptides in lipid bilayers. Using data obtained by PISEMA (Polarization Inversion Spin Exchange at Magic Angle) experiments, periodic spectral patterns arise from well-aligned α-helical molecules. Significant problems in the interpretation of PISEMA spectra may arise for systems that do not form perfectly defined secondary structures, like α-helices, or the signal pattern is disturbed by molecular motion. Here, we present a new method that combines molecular dynamics simulation with tensorial orientational constraints (MDOC) and chemical shift tensor calculations for the simulation and interpretation of PISEMA-like spectra. The calculations include the spectra arising from non α-helical molecules and molecules with non-uniform intrinsic mobility. In a first step, dipolar or quadrupolar interaction tensors drive molecular rotations and reorientations to obtain the proper mean values as observed in corresponding NMR experiments. In a second step, the coordinate snapshots of the MDOC simulations are geometry optimized with the isotropic 15N chemical shifts as constraints using Bond Polarization Theory (BPT) to provide reliable 15N CS tensor data. The averaged dipolar 1H-15N couplings and the δzz tensor components can then be combined to simulate PISEMA patterns. We apply this method to the ß-helical peptide gramicidin A (gA) and demonstrate that this method enables the assignment of most PISEMA resonances. In addition, MDOC simulations provide local order parameters for the calculated sites. These local order parameters reveal large differences in backbone mobility between L- and D-amino acids of gA.
期刊介绍:
MRC is devoted to the rapid publication of papers which are concerned with the development of magnetic resonance techniques, or in which the application of such techniques plays a pivotal part. Contributions from scientists working in all areas of NMR, ESR and NQR are invited, and papers describing applications in all branches of chemistry, structural biology and materials chemistry are published.
The journal is of particular interest not only to scientists working in academic research, but also those working in commercial organisations who need to keep up-to-date with the latest practical applications of magnetic resonance techniques.





 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
