{"title":"zn -卟啉40纳米片的电流密度计算","authors":"Atif Mahmood, Maria Dimitrova and Dage Sundholm*, ","doi":"10.1021/acs.jpca.3c03564","DOIUrl":null,"url":null,"abstract":"<p >Two porphyrinoid nanorings have been studied computationally. They were built by linking 40 Zn-porphyrin units with butadiyne bridges. The molecular structures belonging to the <i>D</i><sub>40<i>h</i></sub> point group were fully optimized with the Turbomole program at the density functional theory (DFT) level using the B3LYP functional and the def2-SVP basis sets. The aromatic character was studied at the DFT level by calculating the magnetically induced current-density (MICD) susceptibility using the GIMIC program. The neutral molecules are globally non-aromatic with aromatic Zn-porphyrin units. Charged nanorings could not be studied because almost degenerate frontier orbitals led to vanishing optical gaps for the cations. Since DFT calculations of the MICD are computationally expensive, we also calculated the MICD using three pseudo-π models. Appropriate pseudo-π models were constructed by removing the outer hydrogen atoms and replacing all carbon and nitrogen atoms with hydrogen atoms. The central Zn atom was either replaced with a beryllium atom or with two inner hydrogen atoms. Calculations with the computationally inexpensive pseudo-π models yielded qualitatively the same magnetic response as obtained in the all-electron calculations.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"127 36","pages":"7452–7459"},"PeriodicalIF":2.8000,"publicationDate":"2023-09-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acs.jpca.3c03564","citationCount":"0","resultStr":"{\"title\":\"Current-Density Calculations on Zn-Porphyrin40 Nanorings\",\"authors\":\"Atif Mahmood, Maria Dimitrova and Dage Sundholm*, \",\"doi\":\"10.1021/acs.jpca.3c03564\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Two porphyrinoid nanorings have been studied computationally. They were built by linking 40 Zn-porphyrin units with butadiyne bridges. The molecular structures belonging to the <i>D</i><sub>40<i>h</i></sub> point group were fully optimized with the Turbomole program at the density functional theory (DFT) level using the B3LYP functional and the def2-SVP basis sets. The aromatic character was studied at the DFT level by calculating the magnetically induced current-density (MICD) susceptibility using the GIMIC program. The neutral molecules are globally non-aromatic with aromatic Zn-porphyrin units. Charged nanorings could not be studied because almost degenerate frontier orbitals led to vanishing optical gaps for the cations. Since DFT calculations of the MICD are computationally expensive, we also calculated the MICD using three pseudo-π models. Appropriate pseudo-π models were constructed by removing the outer hydrogen atoms and replacing all carbon and nitrogen atoms with hydrogen atoms. The central Zn atom was either replaced with a beryllium atom or with two inner hydrogen atoms. Calculations with the computationally inexpensive pseudo-π models yielded qualitatively the same magnetic response as obtained in the all-electron calculations.</p>\",\"PeriodicalId\":59,\"journal\":{\"name\":\"The Journal of Physical Chemistry A\",\"volume\":\"127 36\",\"pages\":\"7452–7459\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2023-09-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.acs.org/doi/epdf/10.1021/acs.jpca.3c03564\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry A\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpca.3c03564\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.3c03564","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Current-Density Calculations on Zn-Porphyrin40 Nanorings
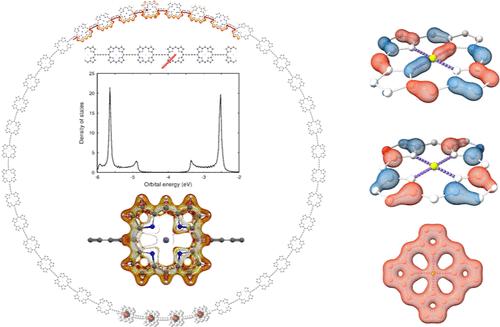
Two porphyrinoid nanorings have been studied computationally. They were built by linking 40 Zn-porphyrin units with butadiyne bridges. The molecular structures belonging to the D40h point group were fully optimized with the Turbomole program at the density functional theory (DFT) level using the B3LYP functional and the def2-SVP basis sets. The aromatic character was studied at the DFT level by calculating the magnetically induced current-density (MICD) susceptibility using the GIMIC program. The neutral molecules are globally non-aromatic with aromatic Zn-porphyrin units. Charged nanorings could not be studied because almost degenerate frontier orbitals led to vanishing optical gaps for the cations. Since DFT calculations of the MICD are computationally expensive, we also calculated the MICD using three pseudo-π models. Appropriate pseudo-π models were constructed by removing the outer hydrogen atoms and replacing all carbon and nitrogen atoms with hydrogen atoms. The central Zn atom was either replaced with a beryllium atom or with two inner hydrogen atoms. Calculations with the computationally inexpensive pseudo-π models yielded qualitatively the same magnetic response as obtained in the all-electron calculations.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
