Ashley M Lane, Jerome T McKay, Herbert L Bonkovsky
{"title":"红细胞原卟啉症的治疗进展——阿麦兰肽的作用。","authors":"Ashley M Lane, Jerome T McKay, Herbert L Bonkovsky","doi":"10.2147/TACG.S122030","DOIUrl":null,"url":null,"abstract":"<p><p>Erythropoietic protoporphyria (EPP) and the phenotypically similar disease X-linked protoporphyria (XLPP) are inherited cutaneous porphyrias characterized clinically by acute non-blistering photosensitivity, intolerance to sunlight, and significantly reduced quality of life. They are due to marked overproduction of protoporphyrin (PP) chiefly by erythroblasts and reticulocytes. In EPP, the underlying genetic defect is in the ferrochelatase gene, which encodes the final enzyme in the heme synthetic pathway. In XLPP, the genetic defect is a gain-of-function mutation, usually a four-base deletion, in the gene that encodes the enzyme 5-aminolevulinic acid synthase-2, the first and rate-controlling enzyme of heme synthesis in developing red blood cells. The excess PP causes acute and painful photosensitivity, being activated by light in the long ultraviolet to blue spectrum (380-420 nm, the Soret band). Although several treatments have been proposed, presently no very effective treatment exists for EPP or XLPP. Afamelanotide (Scenesse<sup>®</sup>) is a first-in-class synthetic analog of α-melanocyte stimulating hormone. Afamelanotide mimics the naturally occurring hormone to increase skin pigmentation by increasing melanin production in melanocytes, resulting in increased sunlight tolerance in those with EPP/XLPP. Afamelanotide is currently approved for use in the European Union and Switzerland, and it is under review in the United States by the Food and Drug Administration for use in patients with EPP/XLPP. This paper provides a review of the clinical characteristics and current therapies for EPP/XLPP. We discuss the pharmacology, clinical efficacy, safety, and tolerability of afamelanotide and summarize the results of several key Phase II and III clinical trials. These data indicate that afamelanotide is a promising therapy for those with these debilitating diseases.</p>","PeriodicalId":39131,"journal":{"name":"Application of Clinical Genetics","volume":"9 1","pages":"179-189"},"PeriodicalIF":2.6000,"publicationDate":"2016-12-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.2147/TACG.S122030","citationCount":"19","resultStr":"{\"title\":\"Advances in the management of erythropoietic protoporphyria - role of afamelanotide.\",\"authors\":\"Ashley M Lane, Jerome T McKay, Herbert L Bonkovsky\",\"doi\":\"10.2147/TACG.S122030\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Erythropoietic protoporphyria (EPP) and the phenotypically similar disease X-linked protoporphyria (XLPP) are inherited cutaneous porphyrias characterized clinically by acute non-blistering photosensitivity, intolerance to sunlight, and significantly reduced quality of life. They are due to marked overproduction of protoporphyrin (PP) chiefly by erythroblasts and reticulocytes. In EPP, the underlying genetic defect is in the ferrochelatase gene, which encodes the final enzyme in the heme synthetic pathway. In XLPP, the genetic defect is a gain-of-function mutation, usually a four-base deletion, in the gene that encodes the enzyme 5-aminolevulinic acid synthase-2, the first and rate-controlling enzyme of heme synthesis in developing red blood cells. The excess PP causes acute and painful photosensitivity, being activated by light in the long ultraviolet to blue spectrum (380-420 nm, the Soret band). Although several treatments have been proposed, presently no very effective treatment exists for EPP or XLPP. Afamelanotide (Scenesse<sup>®</sup>) is a first-in-class synthetic analog of α-melanocyte stimulating hormone. Afamelanotide mimics the naturally occurring hormone to increase skin pigmentation by increasing melanin production in melanocytes, resulting in increased sunlight tolerance in those with EPP/XLPP. Afamelanotide is currently approved for use in the European Union and Switzerland, and it is under review in the United States by the Food and Drug Administration for use in patients with EPP/XLPP. This paper provides a review of the clinical characteristics and current therapies for EPP/XLPP. We discuss the pharmacology, clinical efficacy, safety, and tolerability of afamelanotide and summarize the results of several key Phase II and III clinical trials. These data indicate that afamelanotide is a promising therapy for those with these debilitating diseases.</p>\",\"PeriodicalId\":39131,\"journal\":{\"name\":\"Application of Clinical Genetics\",\"volume\":\"9 1\",\"pages\":\"179-189\"},\"PeriodicalIF\":2.6000,\"publicationDate\":\"2016-12-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.2147/TACG.S122030\",\"citationCount\":\"19\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Application of Clinical Genetics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.2147/TACG.S122030\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2016/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Application of Clinical Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/TACG.S122030","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2016/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Advances in the management of erythropoietic protoporphyria - role of afamelanotide.
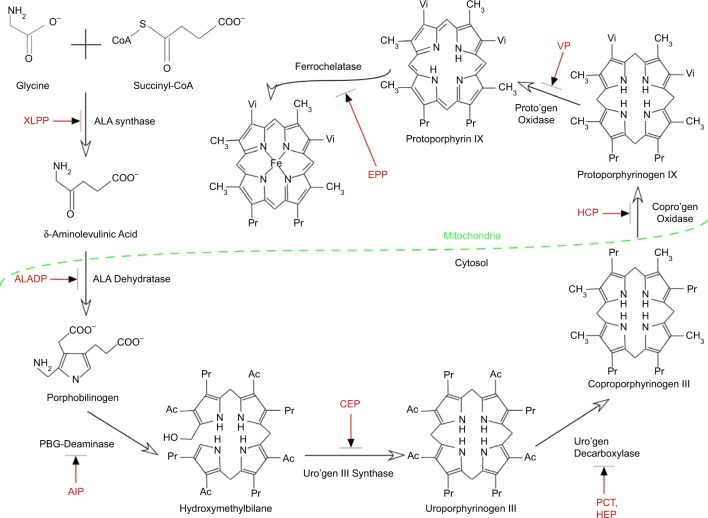
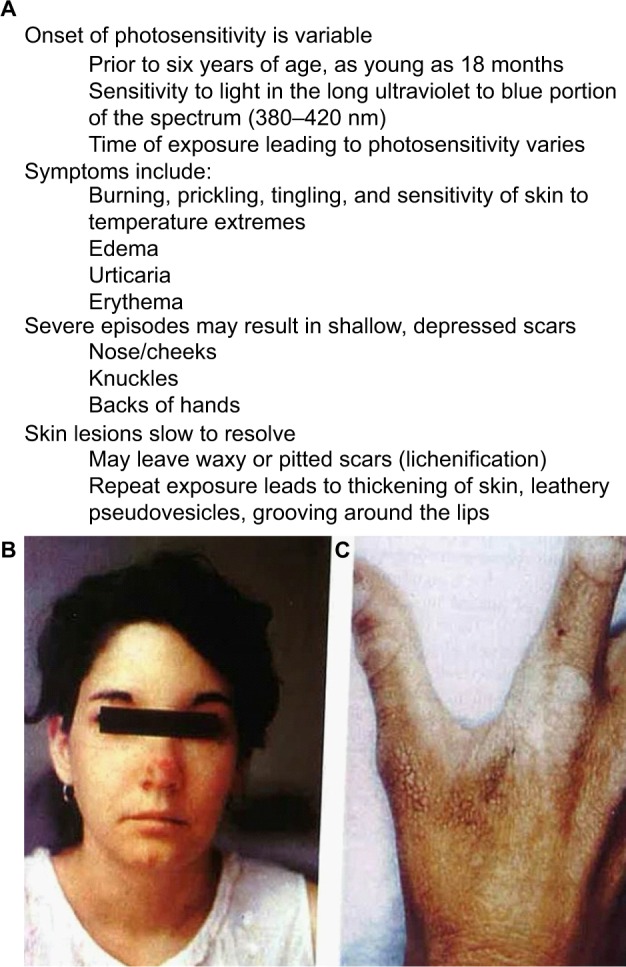
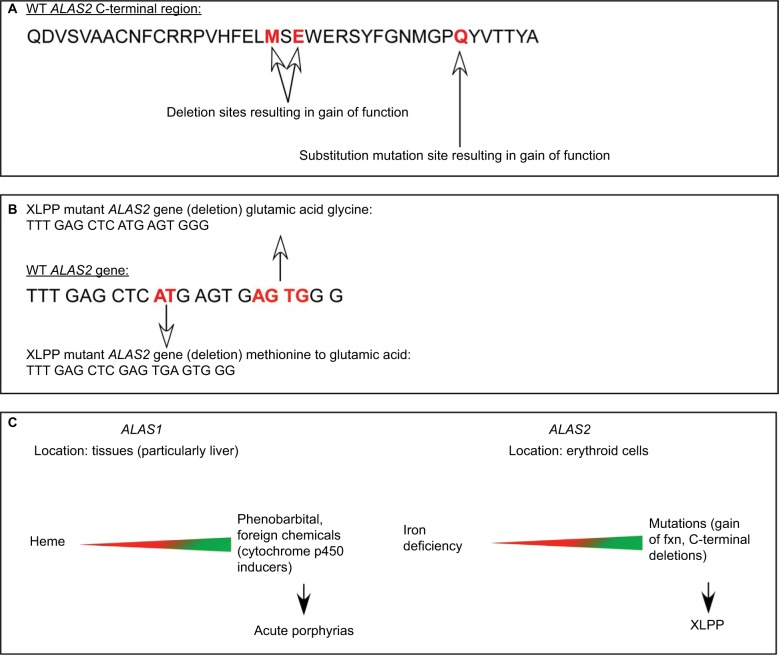
Erythropoietic protoporphyria (EPP) and the phenotypically similar disease X-linked protoporphyria (XLPP) are inherited cutaneous porphyrias characterized clinically by acute non-blistering photosensitivity, intolerance to sunlight, and significantly reduced quality of life. They are due to marked overproduction of protoporphyrin (PP) chiefly by erythroblasts and reticulocytes. In EPP, the underlying genetic defect is in the ferrochelatase gene, which encodes the final enzyme in the heme synthetic pathway. In XLPP, the genetic defect is a gain-of-function mutation, usually a four-base deletion, in the gene that encodes the enzyme 5-aminolevulinic acid synthase-2, the first and rate-controlling enzyme of heme synthesis in developing red blood cells. The excess PP causes acute and painful photosensitivity, being activated by light in the long ultraviolet to blue spectrum (380-420 nm, the Soret band). Although several treatments have been proposed, presently no very effective treatment exists for EPP or XLPP. Afamelanotide (Scenesse®) is a first-in-class synthetic analog of α-melanocyte stimulating hormone. Afamelanotide mimics the naturally occurring hormone to increase skin pigmentation by increasing melanin production in melanocytes, resulting in increased sunlight tolerance in those with EPP/XLPP. Afamelanotide is currently approved for use in the European Union and Switzerland, and it is under review in the United States by the Food and Drug Administration for use in patients with EPP/XLPP. This paper provides a review of the clinical characteristics and current therapies for EPP/XLPP. We discuss the pharmacology, clinical efficacy, safety, and tolerability of afamelanotide and summarize the results of several key Phase II and III clinical trials. These data indicate that afamelanotide is a promising therapy for those with these debilitating diseases.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
