Robert Lersch, Rawan Jannadi, Leonie Grosse, Matias Wagner, Marius Frederik Schneider, Celina von Stülpnagel, Florian Heinen, Heidrun Potschka, Ingo Borggraefe
{"title":"遗传性神经发育障碍的靶向分子策略:Dravet综合征的新经验教训。","authors":"Robert Lersch, Rawan Jannadi, Leonie Grosse, Matias Wagner, Marius Frederik Schneider, Celina von Stülpnagel, Florian Heinen, Heidrun Potschka, Ingo Borggraefe","doi":"10.1177/10738584221088244","DOIUrl":null,"url":null,"abstract":"<p><p>Dravet syndrome is a severe developmental and epileptic encephalopathy mostly caused by heterozygous mutation of the <i>SCN1A</i> gene encoding the voltage-gated sodium channel α subunit Na<sub>v</sub>1.1. Multiple seizure types, cognitive deterioration, behavioral disturbances, ataxia, and sudden unexpected death associated with epilepsy are a hallmark of the disease. Recently approved antiseizure medications such as fenfluramine and cannabidiol have been shown to reduce seizure burden. However, patients with Dravet syndrome are still medically refractory in the majority of cases, and there is a high demand for new therapies aiming to improve behavioral and cognitive outcome. Drug-repurposing approaches for <i>SCN1A</i>-related Dravet syndrome are currently under investigation (i.e., lorcaserin, clemizole, and ataluren). New therapeutic concepts also arise from the field of precision medicine by upregulating functional <i>SCN1A</i> or by activating Na<sub>v</sub>1.1. These include antisense nucleotides directed against the nonproductive transcript of <i>SCN1A</i> with the poison exon 20N and against an inhibitory noncoding antisense RNA of <i>SCN1A</i>. Gene therapy approaches such as adeno-associated virus-based upregulation of <i>SCN1A</i> using a transcriptional activator (ETX101) or CRISPR/dCas technologies show promising results in preclinical studies. Although these new treatment concepts still need further clinical research, they offer great potential for precise and disease modifying treatment of Dravet syndrome.</p>","PeriodicalId":49753,"journal":{"name":"Neuroscientist","volume":"29 6","pages":"732-750"},"PeriodicalIF":3.9000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10623613/pdf/","citationCount":"0","resultStr":"{\"title\":\"Targeted Molecular Strategies for Genetic Neurodevelopmental Disorders: Emerging Lessons from Dravet Syndrome.\",\"authors\":\"Robert Lersch, Rawan Jannadi, Leonie Grosse, Matias Wagner, Marius Frederik Schneider, Celina von Stülpnagel, Florian Heinen, Heidrun Potschka, Ingo Borggraefe\",\"doi\":\"10.1177/10738584221088244\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Dravet syndrome is a severe developmental and epileptic encephalopathy mostly caused by heterozygous mutation of the <i>SCN1A</i> gene encoding the voltage-gated sodium channel α subunit Na<sub>v</sub>1.1. Multiple seizure types, cognitive deterioration, behavioral disturbances, ataxia, and sudden unexpected death associated with epilepsy are a hallmark of the disease. Recently approved antiseizure medications such as fenfluramine and cannabidiol have been shown to reduce seizure burden. However, patients with Dravet syndrome are still medically refractory in the majority of cases, and there is a high demand for new therapies aiming to improve behavioral and cognitive outcome. Drug-repurposing approaches for <i>SCN1A</i>-related Dravet syndrome are currently under investigation (i.e., lorcaserin, clemizole, and ataluren). New therapeutic concepts also arise from the field of precision medicine by upregulating functional <i>SCN1A</i> or by activating Na<sub>v</sub>1.1. These include antisense nucleotides directed against the nonproductive transcript of <i>SCN1A</i> with the poison exon 20N and against an inhibitory noncoding antisense RNA of <i>SCN1A</i>. Gene therapy approaches such as adeno-associated virus-based upregulation of <i>SCN1A</i> using a transcriptional activator (ETX101) or CRISPR/dCas technologies show promising results in preclinical studies. Although these new treatment concepts still need further clinical research, they offer great potential for precise and disease modifying treatment of Dravet syndrome.</p>\",\"PeriodicalId\":49753,\"journal\":{\"name\":\"Neuroscientist\",\"volume\":\"29 6\",\"pages\":\"732-750\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2023-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10623613/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neuroscientist\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1177/10738584221088244\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/4/13 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neuroscientist","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1177/10738584221088244","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/4/13 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Targeted Molecular Strategies for Genetic Neurodevelopmental Disorders: Emerging Lessons from Dravet Syndrome.
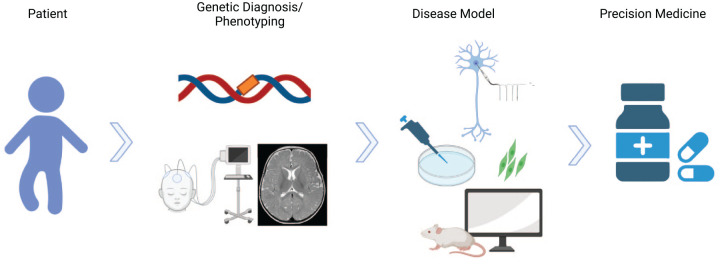
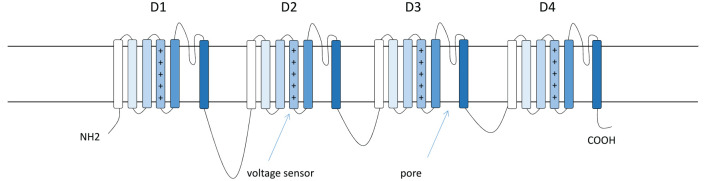
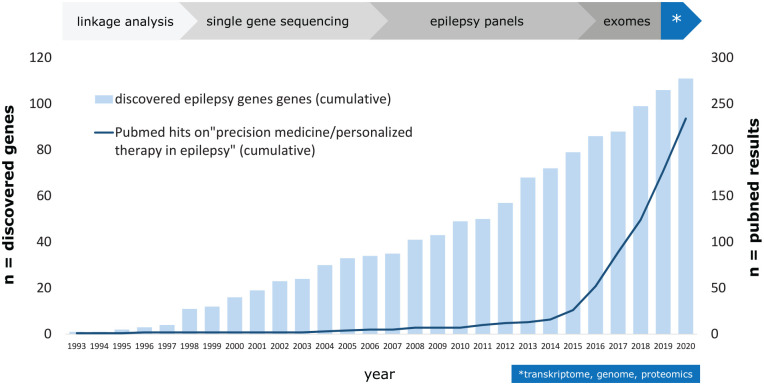
Dravet syndrome is a severe developmental and epileptic encephalopathy mostly caused by heterozygous mutation of the SCN1A gene encoding the voltage-gated sodium channel α subunit Nav1.1. Multiple seizure types, cognitive deterioration, behavioral disturbances, ataxia, and sudden unexpected death associated with epilepsy are a hallmark of the disease. Recently approved antiseizure medications such as fenfluramine and cannabidiol have been shown to reduce seizure burden. However, patients with Dravet syndrome are still medically refractory in the majority of cases, and there is a high demand for new therapies aiming to improve behavioral and cognitive outcome. Drug-repurposing approaches for SCN1A-related Dravet syndrome are currently under investigation (i.e., lorcaserin, clemizole, and ataluren). New therapeutic concepts also arise from the field of precision medicine by upregulating functional SCN1A or by activating Nav1.1. These include antisense nucleotides directed against the nonproductive transcript of SCN1A with the poison exon 20N and against an inhibitory noncoding antisense RNA of SCN1A. Gene therapy approaches such as adeno-associated virus-based upregulation of SCN1A using a transcriptional activator (ETX101) or CRISPR/dCas technologies show promising results in preclinical studies. Although these new treatment concepts still need further clinical research, they offer great potential for precise and disease modifying treatment of Dravet syndrome.
期刊介绍:
Edited by Stephen G. Waxman, The Neuroscientist (NRO) reviews and evaluates the noteworthy advances and key trends in molecular, cellular, developmental, behavioral systems, and cognitive neuroscience in a unique disease-relevant format. Aimed at basic neuroscientists, neurologists, neurosurgeons, and psychiatrists in research, academic, and clinical settings, The Neuroscientist reviews and updates the most important new and emerging basic and clinical neuroscience research.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
