{"title":"基于结构的虚拟筛选和实验策略相结合,从内部数据库中确定咖啡酸酯衍生物作为SARS-CoV-2 3CLpro抑制剂的效力","authors":"Piyatida Pojtanadithee , Kulpornsorn Isswanich , Koonchira Buaban , Supakarn Chamni , Patcharin Wilasluck , Peerapon Deetanya , Kittikhun Wangkanont , Thierry Langer , Peter Wolschann , Kamonpan Sanachai , Thanyada Rungrotmongkol","doi":"10.1016/j.bpc.2023.107125","DOIUrl":null,"url":null,"abstract":"<div><p>Drug development requires significant time and resources, and computer-aided drug discovery techniques that integrate chemical and biological spaces offer valuable tools for the process. This study focused on the field of COVID-19 therapeutics and aimed to identify new active non-covalent inhibitors for 3CL<sup>pro</sup>, a key protein target. By combining <em>in silico</em> and <em>in vitro</em> approaches, an in-house database was utilized to identify potential inhibitors. The drug-likeness criteria were considered to pre-filter 553 compounds from 12 groups of natural products. Using structure-based virtual screening, 296 compounds were identified that matched the chemical features of SARS-CoV-2 3CL<sup>pro</sup> peptidomimetic inhibitor pharmacophore models. Subsequent molecular docking resulted in 43 hits with high binding affinities. Among the hits, caffeic acid analogs showed significant interactions with the 3CL<sup>pro</sup> active site, indicating their potential as promising candidates. To further evaluate their efficacy, enzyme-based assays were conducted, revealing that two ester derivatives of caffeic acid (<strong>4k</strong> and <strong>4l</strong>) exhibited more than a 30% reduction in 3CL<sup>pro</sup> activity. Overall, these findings suggest that the screening approach employed in this study holds promise for the discovery of novel anti-SARS-CoV-2 therapeutics. Furthermore, the methodology could be extended for optimization or retrospective evaluation to enhance molecular targeting and antiviral efficacy of potential drug candidates.</p></div>","PeriodicalId":8979,"journal":{"name":"Biophysical chemistry","volume":"304 ","pages":"Article 107125"},"PeriodicalIF":2.2000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A combination of structure-based virtual screening and experimental strategies to identify the potency of caffeic acid ester derivatives as SARS-CoV-2 3CLpro inhibitor from an in-house database\",\"authors\":\"Piyatida Pojtanadithee , Kulpornsorn Isswanich , Koonchira Buaban , Supakarn Chamni , Patcharin Wilasluck , Peerapon Deetanya , Kittikhun Wangkanont , Thierry Langer , Peter Wolschann , Kamonpan Sanachai , Thanyada Rungrotmongkol\",\"doi\":\"10.1016/j.bpc.2023.107125\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Drug development requires significant time and resources, and computer-aided drug discovery techniques that integrate chemical and biological spaces offer valuable tools for the process. This study focused on the field of COVID-19 therapeutics and aimed to identify new active non-covalent inhibitors for 3CL<sup>pro</sup>, a key protein target. By combining <em>in silico</em> and <em>in vitro</em> approaches, an in-house database was utilized to identify potential inhibitors. The drug-likeness criteria were considered to pre-filter 553 compounds from 12 groups of natural products. Using structure-based virtual screening, 296 compounds were identified that matched the chemical features of SARS-CoV-2 3CL<sup>pro</sup> peptidomimetic inhibitor pharmacophore models. Subsequent molecular docking resulted in 43 hits with high binding affinities. Among the hits, caffeic acid analogs showed significant interactions with the 3CL<sup>pro</sup> active site, indicating their potential as promising candidates. To further evaluate their efficacy, enzyme-based assays were conducted, revealing that two ester derivatives of caffeic acid (<strong>4k</strong> and <strong>4l</strong>) exhibited more than a 30% reduction in 3CL<sup>pro</sup> activity. Overall, these findings suggest that the screening approach employed in this study holds promise for the discovery of novel anti-SARS-CoV-2 therapeutics. Furthermore, the methodology could be extended for optimization or retrospective evaluation to enhance molecular targeting and antiviral efficacy of potential drug candidates.</p></div>\",\"PeriodicalId\":8979,\"journal\":{\"name\":\"Biophysical chemistry\",\"volume\":\"304 \",\"pages\":\"Article 107125\"},\"PeriodicalIF\":2.2000,\"publicationDate\":\"2024-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biophysical chemistry\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S030146222300176X\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/10/20 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biophysical chemistry","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S030146222300176X","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/10/20 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
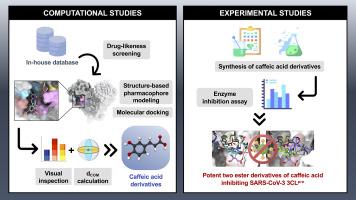
A combination of structure-based virtual screening and experimental strategies to identify the potency of caffeic acid ester derivatives as SARS-CoV-2 3CLpro inhibitor from an in-house database
Drug development requires significant time and resources, and computer-aided drug discovery techniques that integrate chemical and biological spaces offer valuable tools for the process. This study focused on the field of COVID-19 therapeutics and aimed to identify new active non-covalent inhibitors for 3CLpro, a key protein target. By combining in silico and in vitro approaches, an in-house database was utilized to identify potential inhibitors. The drug-likeness criteria were considered to pre-filter 553 compounds from 12 groups of natural products. Using structure-based virtual screening, 296 compounds were identified that matched the chemical features of SARS-CoV-2 3CLpro peptidomimetic inhibitor pharmacophore models. Subsequent molecular docking resulted in 43 hits with high binding affinities. Among the hits, caffeic acid analogs showed significant interactions with the 3CLpro active site, indicating their potential as promising candidates. To further evaluate their efficacy, enzyme-based assays were conducted, revealing that two ester derivatives of caffeic acid (4k and 4l) exhibited more than a 30% reduction in 3CLpro activity. Overall, these findings suggest that the screening approach employed in this study holds promise for the discovery of novel anti-SARS-CoV-2 therapeutics. Furthermore, the methodology could be extended for optimization or retrospective evaluation to enhance molecular targeting and antiviral efficacy of potential drug candidates.
期刊介绍:
Biophysical Chemistry publishes original work and reviews in the areas of chemistry and physics directly impacting biological phenomena. Quantitative analysis of the properties of biological macromolecules, biologically active molecules, macromolecular assemblies and cell components in terms of kinetics, thermodynamics, spatio-temporal organization, NMR and X-ray structural biology, as well as single-molecule detection represent a major focus of the journal. Theoretical and computational treatments of biomacromolecular systems, macromolecular interactions, regulatory control and systems biology are also of interest to the journal.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
