Benjamin Rolles , Andres Caballero-Oteyza , Michele Proietti , Sigune Goldacker , Klaus Warnatz , Nadezhda Camacho-Ordonez , Seraina Prader , Jana Pachlopnik Schmid , Margherita Vieri , Susanne Isfort , Robert Meyer , Martin Kirschner , Tim H. Brümmendorf , Fabian Beier , Bodo Grimbacher
{"title":"端粒生物学障碍可能表现为常见的可变免疫缺陷(CVID)。","authors":"Benjamin Rolles , Andres Caballero-Oteyza , Michele Proietti , Sigune Goldacker , Klaus Warnatz , Nadezhda Camacho-Ordonez , Seraina Prader , Jana Pachlopnik Schmid , Margherita Vieri , Susanne Isfort , Robert Meyer , Martin Kirschner , Tim H. Brümmendorf , Fabian Beier , Bodo Grimbacher","doi":"10.1016/j.clim.2023.109837","DOIUrl":null,"url":null,"abstract":"<div><p>Telomere biology disorders (TBD) are caused by germline pathogenic variants in genes related to telomere maintenance and are characterized by critically short telomeres. In contrast to classical dyskeratosis congenita (DC), which is typically diagnosed in infancy, adult or late onset TBD frequently lack the typical DC triad and rather show variable organ manifestations and a cryptic disease course, thus complicating its diagnosis. Common variable immunodeficiency (CVID), on the other hand, is a primary antibody deficiency (PAD) syndrome. PADs are a heterogenous group of diseases characterized by hypogammaglobulinemia which occurs due to dysfunctional B lymphocytes and additional autoimmune and autoinflammatory complications. Genetic screening reveals a monogenic cause in a subset of CVID patients (15–35%). In our study, we screened the exomes of 491 CVID patients for the occurrence of TBD-related variants in 13 genes encoding for telomere/telomerase-associated proteins, which had previously been linked to the disease. We found 110/491 patients (22%) carrying 91 rare candidate variants in these 13 genes. Following the American College of Medical Genetics and Genomics (ACMG) guidelines, we classified two variants as benign, two as likely benign, 64 as variants of uncertain significance (VUS), four as likely pathogenic, and one heterozygous variant in an autosomal recessive disease gene as pathogenic. We performed telomere length measurement in 42 of the 110 patients with candidate variants and CVID. Two of these 42 patients showed significantly shorter telomeres compared to controls in both lymphocytes and granulocytes. Following the evaluation of the published literature and the patient's manifestations, we re-classified two VUS as likely pathogenic variants. Thus, 0.5–1% of all CVID patients in our study carry possibly pathogenic variants in telomere/telomerase-associated genes. Our data adds CVID to the broad clinical spectrum of cryptic adult-onset TBD. As the molecular diagnosis greatly impacts patient management and treatment strategies, we advise inclusion of all TBD-associated genes—despite their low prevalence—into the molecular screening of patients with antibody deficiencies.</p></div>","PeriodicalId":10392,"journal":{"name":"Clinical immunology","volume":"257 ","pages":"Article 109837"},"PeriodicalIF":3.8000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Telomere biology disorders may manifest as common variable immunodeficiency (CVID)\",\"authors\":\"Benjamin Rolles , Andres Caballero-Oteyza , Michele Proietti , Sigune Goldacker , Klaus Warnatz , Nadezhda Camacho-Ordonez , Seraina Prader , Jana Pachlopnik Schmid , Margherita Vieri , Susanne Isfort , Robert Meyer , Martin Kirschner , Tim H. Brümmendorf , Fabian Beier , Bodo Grimbacher\",\"doi\":\"10.1016/j.clim.2023.109837\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Telomere biology disorders (TBD) are caused by germline pathogenic variants in genes related to telomere maintenance and are characterized by critically short telomeres. In contrast to classical dyskeratosis congenita (DC), which is typically diagnosed in infancy, adult or late onset TBD frequently lack the typical DC triad and rather show variable organ manifestations and a cryptic disease course, thus complicating its diagnosis. Common variable immunodeficiency (CVID), on the other hand, is a primary antibody deficiency (PAD) syndrome. PADs are a heterogenous group of diseases characterized by hypogammaglobulinemia which occurs due to dysfunctional B lymphocytes and additional autoimmune and autoinflammatory complications. Genetic screening reveals a monogenic cause in a subset of CVID patients (15–35%). In our study, we screened the exomes of 491 CVID patients for the occurrence of TBD-related variants in 13 genes encoding for telomere/telomerase-associated proteins, which had previously been linked to the disease. We found 110/491 patients (22%) carrying 91 rare candidate variants in these 13 genes. Following the American College of Medical Genetics and Genomics (ACMG) guidelines, we classified two variants as benign, two as likely benign, 64 as variants of uncertain significance (VUS), four as likely pathogenic, and one heterozygous variant in an autosomal recessive disease gene as pathogenic. We performed telomere length measurement in 42 of the 110 patients with candidate variants and CVID. Two of these 42 patients showed significantly shorter telomeres compared to controls in both lymphocytes and granulocytes. Following the evaluation of the published literature and the patient's manifestations, we re-classified two VUS as likely pathogenic variants. Thus, 0.5–1% of all CVID patients in our study carry possibly pathogenic variants in telomere/telomerase-associated genes. Our data adds CVID to the broad clinical spectrum of cryptic adult-onset TBD. As the molecular diagnosis greatly impacts patient management and treatment strategies, we advise inclusion of all TBD-associated genes—despite their low prevalence—into the molecular screening of patients with antibody deficiencies.</p></div>\",\"PeriodicalId\":10392,\"journal\":{\"name\":\"Clinical immunology\",\"volume\":\"257 \",\"pages\":\"Article 109837\"},\"PeriodicalIF\":3.8000,\"publicationDate\":\"2023-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical immunology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1521661623006009\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/11/8 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"IMMUNOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical immunology","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1521661623006009","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/11/8 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
Telomere biology disorders may manifest as common variable immunodeficiency (CVID)
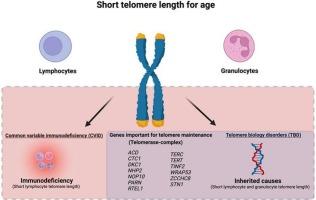
Telomere biology disorders (TBD) are caused by germline pathogenic variants in genes related to telomere maintenance and are characterized by critically short telomeres. In contrast to classical dyskeratosis congenita (DC), which is typically diagnosed in infancy, adult or late onset TBD frequently lack the typical DC triad and rather show variable organ manifestations and a cryptic disease course, thus complicating its diagnosis. Common variable immunodeficiency (CVID), on the other hand, is a primary antibody deficiency (PAD) syndrome. PADs are a heterogenous group of diseases characterized by hypogammaglobulinemia which occurs due to dysfunctional B lymphocytes and additional autoimmune and autoinflammatory complications. Genetic screening reveals a monogenic cause in a subset of CVID patients (15–35%). In our study, we screened the exomes of 491 CVID patients for the occurrence of TBD-related variants in 13 genes encoding for telomere/telomerase-associated proteins, which had previously been linked to the disease. We found 110/491 patients (22%) carrying 91 rare candidate variants in these 13 genes. Following the American College of Medical Genetics and Genomics (ACMG) guidelines, we classified two variants as benign, two as likely benign, 64 as variants of uncertain significance (VUS), four as likely pathogenic, and one heterozygous variant in an autosomal recessive disease gene as pathogenic. We performed telomere length measurement in 42 of the 110 patients with candidate variants and CVID. Two of these 42 patients showed significantly shorter telomeres compared to controls in both lymphocytes and granulocytes. Following the evaluation of the published literature and the patient's manifestations, we re-classified two VUS as likely pathogenic variants. Thus, 0.5–1% of all CVID patients in our study carry possibly pathogenic variants in telomere/telomerase-associated genes. Our data adds CVID to the broad clinical spectrum of cryptic adult-onset TBD. As the molecular diagnosis greatly impacts patient management and treatment strategies, we advise inclusion of all TBD-associated genes—despite their low prevalence—into the molecular screening of patients with antibody deficiencies.
期刊介绍:
Clinical Immunology publishes original research delving into the molecular and cellular foundations of immunological diseases. Additionally, the journal includes reviews covering timely subjects in basic immunology, along with case reports and letters to the editor.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
