Luyao Hao, Wenjin Chen, X. Lei, Wenjing Yao, Nan Wang
{"title":"γ - Ni3Nb低折射率表面的结构、能量和电子性质:第一性原理计算","authors":"Luyao Hao, Wenjin Chen, X. Lei, Wenjing Yao, Nan Wang","doi":"10.1002/pssb.202300239","DOIUrl":null,"url":null,"abstract":"First‐principles calculations are carried out to study the surface structure, energies, and electronic properties of Ni3Nb(100), Ni3Nb(001), and Ni3Nb(110) based on the density functional theory (DFT). The surface relaxation results reveal that the relaxations are mainly localized in the first and second atomic layer, and Ni3Nb(110)‐Ni experiences the largest surface relaxation (–16.95%), whereas Ni3Nb(001)‐NiNb undergoes smallest relaxations. The surface energies of nonstoichiometric surfaces present a linear relationship with the chemical potential of Ni (ΔμNi), while those of stoichiometric surface are independent of ΔμNi. Furthermore, Ni3Nb(001)–Ni and Ni3Nb(001)–NiNb are the most stable surfaces owing to their having the lowest surface energy in a wide range of ΔμNi, while the nonstoichiometric Ni3Nb(110)–Ni and Ni3Nb(110)–NiNb surfaces with the largest surface energies are the most unstable surfaces. The electronic structures of nonstoichiometric surfaces are different from that of the bulk Ni3Nb, whereas the effect of surface relaxation on the electronic properties of the stoichiometric surface is weak.","PeriodicalId":20107,"journal":{"name":"physica status solidi (b)","volume":"74 1","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2023-09-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Structures, energies, and electronic properties of low‐index surfaces of γꞌꞌ‐Ni3Nb: A first‐principles calculations\",\"authors\":\"Luyao Hao, Wenjin Chen, X. Lei, Wenjing Yao, Nan Wang\",\"doi\":\"10.1002/pssb.202300239\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"First‐principles calculations are carried out to study the surface structure, energies, and electronic properties of Ni3Nb(100), Ni3Nb(001), and Ni3Nb(110) based on the density functional theory (DFT). The surface relaxation results reveal that the relaxations are mainly localized in the first and second atomic layer, and Ni3Nb(110)‐Ni experiences the largest surface relaxation (–16.95%), whereas Ni3Nb(001)‐NiNb undergoes smallest relaxations. The surface energies of nonstoichiometric surfaces present a linear relationship with the chemical potential of Ni (ΔμNi), while those of stoichiometric surface are independent of ΔμNi. Furthermore, Ni3Nb(001)–Ni and Ni3Nb(001)–NiNb are the most stable surfaces owing to their having the lowest surface energy in a wide range of ΔμNi, while the nonstoichiometric Ni3Nb(110)–Ni and Ni3Nb(110)–NiNb surfaces with the largest surface energies are the most unstable surfaces. The electronic structures of nonstoichiometric surfaces are different from that of the bulk Ni3Nb, whereas the effect of surface relaxation on the electronic properties of the stoichiometric surface is weak.\",\"PeriodicalId\":20107,\"journal\":{\"name\":\"physica status solidi (b)\",\"volume\":\"74 1\",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-09-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"physica status solidi (b)\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1002/pssb.202300239\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"physica status solidi (b)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1002/pssb.202300239","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Structures, energies, and electronic properties of low‐index surfaces of γꞌꞌ‐Ni3Nb: A first‐principles calculations
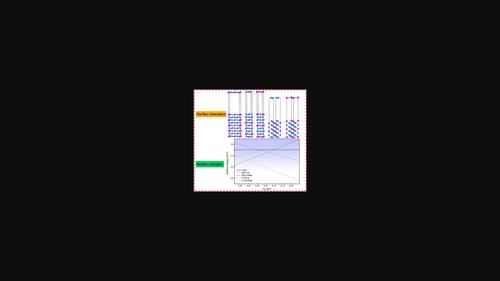
First‐principles calculations are carried out to study the surface structure, energies, and electronic properties of Ni3Nb(100), Ni3Nb(001), and Ni3Nb(110) based on the density functional theory (DFT). The surface relaxation results reveal that the relaxations are mainly localized in the first and second atomic layer, and Ni3Nb(110)‐Ni experiences the largest surface relaxation (–16.95%), whereas Ni3Nb(001)‐NiNb undergoes smallest relaxations. The surface energies of nonstoichiometric surfaces present a linear relationship with the chemical potential of Ni (ΔμNi), while those of stoichiometric surface are independent of ΔμNi. Furthermore, Ni3Nb(001)–Ni and Ni3Nb(001)–NiNb are the most stable surfaces owing to their having the lowest surface energy in a wide range of ΔμNi, while the nonstoichiometric Ni3Nb(110)–Ni and Ni3Nb(110)–NiNb surfaces with the largest surface energies are the most unstable surfaces. The electronic structures of nonstoichiometric surfaces are different from that of the bulk Ni3Nb, whereas the effect of surface relaxation on the electronic properties of the stoichiometric surface is weak.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
