Jáchym Pavliš, Alex Mathers, Michal Fulem and Martin Klajmon*,
{"title":"PC-SAFT活度系数的纯预测是否有助于药物-聚合物相容性筛选?","authors":"Jáchym Pavliš, Alex Mathers, Michal Fulem and Martin Klajmon*, ","doi":"10.1021/acs.molpharmaceut.3c00124","DOIUrl":null,"url":null,"abstract":"<p >The bioavailability of poorly water-soluble active pharmaceutical ingredients (APIs) can be improved <i>via</i> the formulation of an amorphous solid dispersion (ASD), where the API is incorporated into a suitable polymeric carrier. Optimal carriers that exhibit good compatibility (i.e., solubility and miscibility) with given APIs are typically identified through experimental means, which are routinely labor- and cost-inefficient. Therefore, the perturbed-chain statistical associating fluid theory (PC-SAFT) equation of state, a popular thermodynamic model in pharmaceutical applications, is examined in terms of its performance regarding the computational pure prediction of API–polymer compatibility based on activity coefficients (API fusion properties were taken from experiments) without any binary interaction parameters fitted to API–polymer experimental data (that is, <i>k</i><sub><i>ij</i></sub> = 0 in all cases). This kind of prediction does not need any experimental binary information and has been underreported in the literature so far, as the routine modeling strategy used in the majority of the existing PC-SAFT applications to ASDs comprised the use of nonzero <i>k</i><sub><i>ij</i></sub> values. The predictive performance of PC-SAFT was systematically and thoroughly evaluated against reliable experimental data for almost 40 API–polymer combinations. We also examined the effect of different sets of PC-SAFT parameters for APIs on compatibility predictions. Quantitatively, the total average error calculated over all systems was approximately 50% in the weight fraction solubility of APIs in polymers, regardless of the specific API parametrization. The magnitude of the error for individual systems was found to vary significantly from one system to another. Interestingly, the poorest results were obtained for systems with self-associating polymers such as poly(vinyl alcohol). Such polymers can form intramolecular hydrogen bonds, which are not accounted for in the PC-SAFT variant routinely applied to ASDs (i.e., that used in this work). However, the qualitative ranking of polymers with respect to their compatibility with a given API was reasonably predicted in many cases. It was also predicted correctly that some polymers always have better compatibility with the APIs than others. Finally, possible future routes to improve the cost–performance ratio of PC-SAFT in terms of parametrization are discussed.</p>","PeriodicalId":52,"journal":{"name":"Molecular Pharmaceutics","volume":"20 8","pages":"3960–3974"},"PeriodicalIF":4.5000,"publicationDate":"2023-06-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acs.molpharmaceut.3c00124","citationCount":"1","resultStr":"{\"title\":\"Can Pure Predictions of Activity Coefficients from PC-SAFT Assist Drug–Polymer Compatibility Screening?\",\"authors\":\"Jáchym Pavliš, Alex Mathers, Michal Fulem and Martin Klajmon*, \",\"doi\":\"10.1021/acs.molpharmaceut.3c00124\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The bioavailability of poorly water-soluble active pharmaceutical ingredients (APIs) can be improved <i>via</i> the formulation of an amorphous solid dispersion (ASD), where the API is incorporated into a suitable polymeric carrier. Optimal carriers that exhibit good compatibility (i.e., solubility and miscibility) with given APIs are typically identified through experimental means, which are routinely labor- and cost-inefficient. Therefore, the perturbed-chain statistical associating fluid theory (PC-SAFT) equation of state, a popular thermodynamic model in pharmaceutical applications, is examined in terms of its performance regarding the computational pure prediction of API–polymer compatibility based on activity coefficients (API fusion properties were taken from experiments) without any binary interaction parameters fitted to API–polymer experimental data (that is, <i>k</i><sub><i>ij</i></sub> = 0 in all cases). This kind of prediction does not need any experimental binary information and has been underreported in the literature so far, as the routine modeling strategy used in the majority of the existing PC-SAFT applications to ASDs comprised the use of nonzero <i>k</i><sub><i>ij</i></sub> values. The predictive performance of PC-SAFT was systematically and thoroughly evaluated against reliable experimental data for almost 40 API–polymer combinations. We also examined the effect of different sets of PC-SAFT parameters for APIs on compatibility predictions. Quantitatively, the total average error calculated over all systems was approximately 50% in the weight fraction solubility of APIs in polymers, regardless of the specific API parametrization. The magnitude of the error for individual systems was found to vary significantly from one system to another. Interestingly, the poorest results were obtained for systems with self-associating polymers such as poly(vinyl alcohol). Such polymers can form intramolecular hydrogen bonds, which are not accounted for in the PC-SAFT variant routinely applied to ASDs (i.e., that used in this work). However, the qualitative ranking of polymers with respect to their compatibility with a given API was reasonably predicted in many cases. It was also predicted correctly that some polymers always have better compatibility with the APIs than others. Finally, possible future routes to improve the cost–performance ratio of PC-SAFT in terms of parametrization are discussed.</p>\",\"PeriodicalId\":52,\"journal\":{\"name\":\"Molecular Pharmaceutics\",\"volume\":\"20 8\",\"pages\":\"3960–3974\"},\"PeriodicalIF\":4.5000,\"publicationDate\":\"2023-06-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.acs.org/doi/epdf/10.1021/acs.molpharmaceut.3c00124\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Pharmaceutics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.3c00124\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"MEDICINE, RESEARCH & EXPERIMENTAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Pharmaceutics","FirstCategoryId":"3","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.3c00124","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
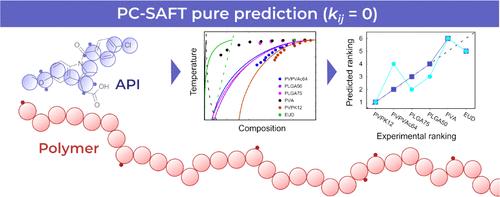
Can Pure Predictions of Activity Coefficients from PC-SAFT Assist Drug–Polymer Compatibility Screening?
The bioavailability of poorly water-soluble active pharmaceutical ingredients (APIs) can be improved via the formulation of an amorphous solid dispersion (ASD), where the API is incorporated into a suitable polymeric carrier. Optimal carriers that exhibit good compatibility (i.e., solubility and miscibility) with given APIs are typically identified through experimental means, which are routinely labor- and cost-inefficient. Therefore, the perturbed-chain statistical associating fluid theory (PC-SAFT) equation of state, a popular thermodynamic model in pharmaceutical applications, is examined in terms of its performance regarding the computational pure prediction of API–polymer compatibility based on activity coefficients (API fusion properties were taken from experiments) without any binary interaction parameters fitted to API–polymer experimental data (that is, kij = 0 in all cases). This kind of prediction does not need any experimental binary information and has been underreported in the literature so far, as the routine modeling strategy used in the majority of the existing PC-SAFT applications to ASDs comprised the use of nonzero kij values. The predictive performance of PC-SAFT was systematically and thoroughly evaluated against reliable experimental data for almost 40 API–polymer combinations. We also examined the effect of different sets of PC-SAFT parameters for APIs on compatibility predictions. Quantitatively, the total average error calculated over all systems was approximately 50% in the weight fraction solubility of APIs in polymers, regardless of the specific API parametrization. The magnitude of the error for individual systems was found to vary significantly from one system to another. Interestingly, the poorest results were obtained for systems with self-associating polymers such as poly(vinyl alcohol). Such polymers can form intramolecular hydrogen bonds, which are not accounted for in the PC-SAFT variant routinely applied to ASDs (i.e., that used in this work). However, the qualitative ranking of polymers with respect to their compatibility with a given API was reasonably predicted in many cases. It was also predicted correctly that some polymers always have better compatibility with the APIs than others. Finally, possible future routes to improve the cost–performance ratio of PC-SAFT in terms of parametrization are discussed.
期刊介绍:
Molecular Pharmaceutics publishes the results of original research that contributes significantly to the molecular mechanistic understanding of drug delivery and drug delivery systems. The journal encourages contributions describing research at the interface of drug discovery and drug development.
Scientific areas within the scope of the journal include physical and pharmaceutical chemistry, biochemistry and biophysics, molecular and cellular biology, and polymer and materials science as they relate to drug and drug delivery system efficacy. Mechanistic Drug Delivery and Drug Targeting research on modulating activity and efficacy of a drug or drug product is within the scope of Molecular Pharmaceutics. Theoretical and experimental peer-reviewed research articles, communications, reviews, and perspectives are welcomed.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
