Sameer Bahal, Maha E Houssen, Ania Manson, Lorena Lorenzo, Mark A Russell, Noel G Morgan, Fariba Tahami, Sofia Grigoriadou
{"title":"STAT3突变导致高IgE综合征导致转录活性抑制的证据。","authors":"Sameer Bahal, Maha E Houssen, Ania Manson, Lorena Lorenzo, Mark A Russell, Noel G Morgan, Fariba Tahami, Sofia Grigoriadou","doi":"10.1155/2019/1869524","DOIUrl":null,"url":null,"abstract":"<p><p>We present the case of a 19-year-old female with a mild form of Autosomal Dominant Hyper IgE syndrome (HIES) associated with a loss-of-function mutation in <i>STAT3</i>. Within the first years of life she developed multiple, <i>Staphylococcus aureus</i> associated abscesses in the neck and face requiring frequent incision and drainage. Respiratory tract infections were not a feature of the clinical phenotype and a high resolution thoracic CT scan was unremarkable. Retained dentition was noted but fungal nail disease and recurrent thrush were absent. The total IgE was 970 IU/L, Lymphocyte counts and immunoglobulin levels were normal (IgG borderline 18.5 gr/L). There was suboptimal response to test immunisation with Pneumovax II vaccine. Th17 cell phenotyping revealed low levels of IL-17 expressing cells (0.3% of total CD4 T Cells numbers). Genetic analysis identified a missense mutation, N567D, in a conserved region of the linker domain of STAT3. Functional studies in HEK293 cells reveal that this mutation potently inhibits STAT3 activity when compared to the wildtype protein. This is consistent with other reported mutations in <i>STAT3</i> associated with HIES. However, surprisingly, the magnitude of inhibition was similar to another STAT3 mutation (V637M) which causes a much more severe form of the disease.</p>","PeriodicalId":42865,"journal":{"name":"Case Reports in Immunology","volume":"2019 ","pages":"1869524"},"PeriodicalIF":1.5000,"publicationDate":"2019-10-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6815622/pdf/","citationCount":"0","resultStr":"{\"title\":\"Evidence that a STAT3 Mutation Causing Hyper IgE Syndrome Leads to Repression of Transcriptional Activity.\",\"authors\":\"Sameer Bahal, Maha E Houssen, Ania Manson, Lorena Lorenzo, Mark A Russell, Noel G Morgan, Fariba Tahami, Sofia Grigoriadou\",\"doi\":\"10.1155/2019/1869524\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>We present the case of a 19-year-old female with a mild form of Autosomal Dominant Hyper IgE syndrome (HIES) associated with a loss-of-function mutation in <i>STAT3</i>. Within the first years of life she developed multiple, <i>Staphylococcus aureus</i> associated abscesses in the neck and face requiring frequent incision and drainage. Respiratory tract infections were not a feature of the clinical phenotype and a high resolution thoracic CT scan was unremarkable. Retained dentition was noted but fungal nail disease and recurrent thrush were absent. The total IgE was 970 IU/L, Lymphocyte counts and immunoglobulin levels were normal (IgG borderline 18.5 gr/L). There was suboptimal response to test immunisation with Pneumovax II vaccine. Th17 cell phenotyping revealed low levels of IL-17 expressing cells (0.3% of total CD4 T Cells numbers). Genetic analysis identified a missense mutation, N567D, in a conserved region of the linker domain of STAT3. Functional studies in HEK293 cells reveal that this mutation potently inhibits STAT3 activity when compared to the wildtype protein. This is consistent with other reported mutations in <i>STAT3</i> associated with HIES. However, surprisingly, the magnitude of inhibition was similar to another STAT3 mutation (V637M) which causes a much more severe form of the disease.</p>\",\"PeriodicalId\":42865,\"journal\":{\"name\":\"Case Reports in Immunology\",\"volume\":\"2019 \",\"pages\":\"1869524\"},\"PeriodicalIF\":1.5000,\"publicationDate\":\"2019-10-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6815622/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Case Reports in Immunology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2019/1869524\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2019/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q4\",\"JCRName\":\"IMMUNOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Immunology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2019/1869524","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2019/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
Evidence that a STAT3 Mutation Causing Hyper IgE Syndrome Leads to Repression of Transcriptional Activity.
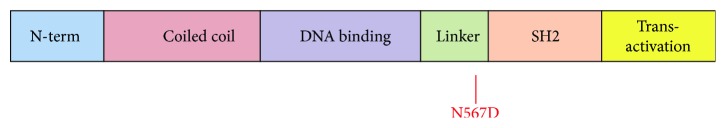
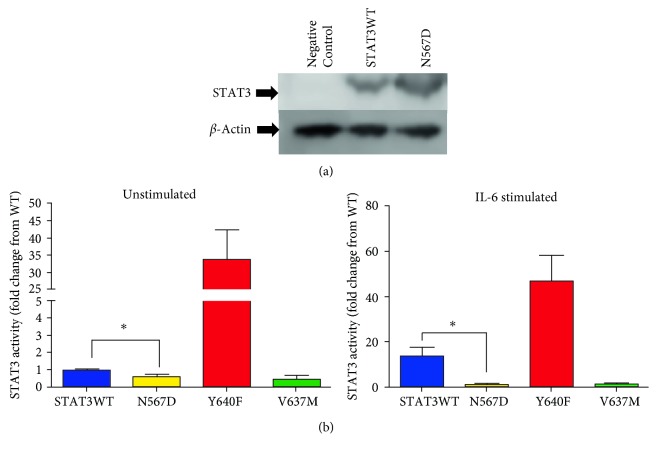
We present the case of a 19-year-old female with a mild form of Autosomal Dominant Hyper IgE syndrome (HIES) associated with a loss-of-function mutation in STAT3. Within the first years of life she developed multiple, Staphylococcus aureus associated abscesses in the neck and face requiring frequent incision and drainage. Respiratory tract infections were not a feature of the clinical phenotype and a high resolution thoracic CT scan was unremarkable. Retained dentition was noted but fungal nail disease and recurrent thrush were absent. The total IgE was 970 IU/L, Lymphocyte counts and immunoglobulin levels were normal (IgG borderline 18.5 gr/L). There was suboptimal response to test immunisation with Pneumovax II vaccine. Th17 cell phenotyping revealed low levels of IL-17 expressing cells (0.3% of total CD4 T Cells numbers). Genetic analysis identified a missense mutation, N567D, in a conserved region of the linker domain of STAT3. Functional studies in HEK293 cells reveal that this mutation potently inhibits STAT3 activity when compared to the wildtype protein. This is consistent with other reported mutations in STAT3 associated with HIES. However, surprisingly, the magnitude of inhibition was similar to another STAT3 mutation (V637M) which causes a much more severe form of the disease.
期刊介绍:
Case Reports in Immunology is a peer-reviewed, Open Access journal that publishes case reports and case series related to allergies, immunodeficiencies, autoimmune diseases, immune disorders, cancer immunology and transplantation immunology.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
