Payal Jain, Sudarshan Iyer, Joshua Straka, Lea F Surrey, Jennifer Pogoriler, Harry Han, Tiffany Smith, Christine Busch, Elizabeth Fox, Marilyn Li, Angela J Waanders, Adam Resnick, Monika A Davare
{"title":"一种新的NOTCH1-ROS1基因融合在儿童血管肉瘤中的致癌性和靶向性的发现和功能特征","authors":"Payal Jain, Sudarshan Iyer, Joshua Straka, Lea F Surrey, Jennifer Pogoriler, Harry Han, Tiffany Smith, Christine Busch, Elizabeth Fox, Marilyn Li, Angela J Waanders, Adam Resnick, Monika A Davare","doi":"10.1101/mcs.a006222","DOIUrl":null,"url":null,"abstract":"<p><p>Angiosarcomas are rare, malignant soft tissue tumors in children that arise in a wide range of anatomical locations and have limited targeted therapies available. Here, we report a rare case of a pediatric angiosarcoma (pAS) with Li-Fraumeni syndrome (LFS) expressing a novel <i>NOTCH1-ROS1</i> gene fusion. Although both <i>NOTCH1</i> and <i>ROS1</i> are established proto-oncogenes, our study is the first to describe the mechanistic role of <i>NOTCH1</i>-<i>ROS1</i> fusion arising via intrachromosomal rearrangement. NOTCH1-ROS1 displayed potent neoplastic transformation propensity in vitro, and harbors tumorigenic potential in vivo, where it induced oncogenic activation of the MAPK, PI3K/mTOR, and JAK-STAT signaling pathways in a murine allograft model. We found an unexpected contribution of the NOTCH1 extracellular region in mediating NOTCH1-ROS1 activation and oncogenic function, highlighting the contribution of both NOTCH1 and ROS1 fusion partners in driving tumorigenicity. Interestingly, neither membrane localization nor fusion protein dimerization were found to be essential for NOTCH1-ROS1 fusion oncogenicity. To target NOTCH1-ROS1-driven tumors, we tested both NOTCH1-directed inhibitors and ROS1-targeted tyrosine kinase inhibitors (TKI) in heterologous models (NIH3T3, Ba/F3). Although NOTCH1 inhibitors did not suppress NOTCH1-ROS1-driven oncogenic growth, we found that oral entrectinib treatment effectively suppressed the growth of NOTCH-ROS1-driven tumors. Taken together, we report the first known pAS case with a novel NOTCH1-ROS1 alteration along with a detailed report on the function and therapeutic targeting of NOTCH1-ROS1. Our study highlights the importance of genomic profiling of rare cancers such as pAS to reveal actionable drivers and improve patient outcomes.</p>","PeriodicalId":10360,"journal":{"name":"Cold Spring Harbor Molecular Case Studies","volume":"8 6","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2022-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/d0/65/MCS006222Jai.PMC9632357.pdf","citationCount":"1","resultStr":"{\"title\":\"Discovery and functional characterization of the oncogenicity and targetability of a novel <i>NOTCH1-ROS1</i> gene fusion in pediatric angiosarcoma.\",\"authors\":\"Payal Jain, Sudarshan Iyer, Joshua Straka, Lea F Surrey, Jennifer Pogoriler, Harry Han, Tiffany Smith, Christine Busch, Elizabeth Fox, Marilyn Li, Angela J Waanders, Adam Resnick, Monika A Davare\",\"doi\":\"10.1101/mcs.a006222\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Angiosarcomas are rare, malignant soft tissue tumors in children that arise in a wide range of anatomical locations and have limited targeted therapies available. Here, we report a rare case of a pediatric angiosarcoma (pAS) with Li-Fraumeni syndrome (LFS) expressing a novel <i>NOTCH1-ROS1</i> gene fusion. Although both <i>NOTCH1</i> and <i>ROS1</i> are established proto-oncogenes, our study is the first to describe the mechanistic role of <i>NOTCH1</i>-<i>ROS1</i> fusion arising via intrachromosomal rearrangement. NOTCH1-ROS1 displayed potent neoplastic transformation propensity in vitro, and harbors tumorigenic potential in vivo, where it induced oncogenic activation of the MAPK, PI3K/mTOR, and JAK-STAT signaling pathways in a murine allograft model. We found an unexpected contribution of the NOTCH1 extracellular region in mediating NOTCH1-ROS1 activation and oncogenic function, highlighting the contribution of both NOTCH1 and ROS1 fusion partners in driving tumorigenicity. Interestingly, neither membrane localization nor fusion protein dimerization were found to be essential for NOTCH1-ROS1 fusion oncogenicity. To target NOTCH1-ROS1-driven tumors, we tested both NOTCH1-directed inhibitors and ROS1-targeted tyrosine kinase inhibitors (TKI) in heterologous models (NIH3T3, Ba/F3). Although NOTCH1 inhibitors did not suppress NOTCH1-ROS1-driven oncogenic growth, we found that oral entrectinib treatment effectively suppressed the growth of NOTCH-ROS1-driven tumors. Taken together, we report the first known pAS case with a novel NOTCH1-ROS1 alteration along with a detailed report on the function and therapeutic targeting of NOTCH1-ROS1. Our study highlights the importance of genomic profiling of rare cancers such as pAS to reveal actionable drivers and improve patient outcomes.</p>\",\"PeriodicalId\":10360,\"journal\":{\"name\":\"Cold Spring Harbor Molecular Case Studies\",\"volume\":\"8 6\",\"pages\":\"\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2022-10-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/d0/65/MCS006222Jai.PMC9632357.pdf\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cold Spring Harbor Molecular Case Studies\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1101/mcs.a006222\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"MEDICINE, RESEARCH & EXPERIMENTAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cold Spring Harbor Molecular Case Studies","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/mcs.a006222","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
Discovery and functional characterization of the oncogenicity and targetability of a novel NOTCH1-ROS1 gene fusion in pediatric angiosarcoma.
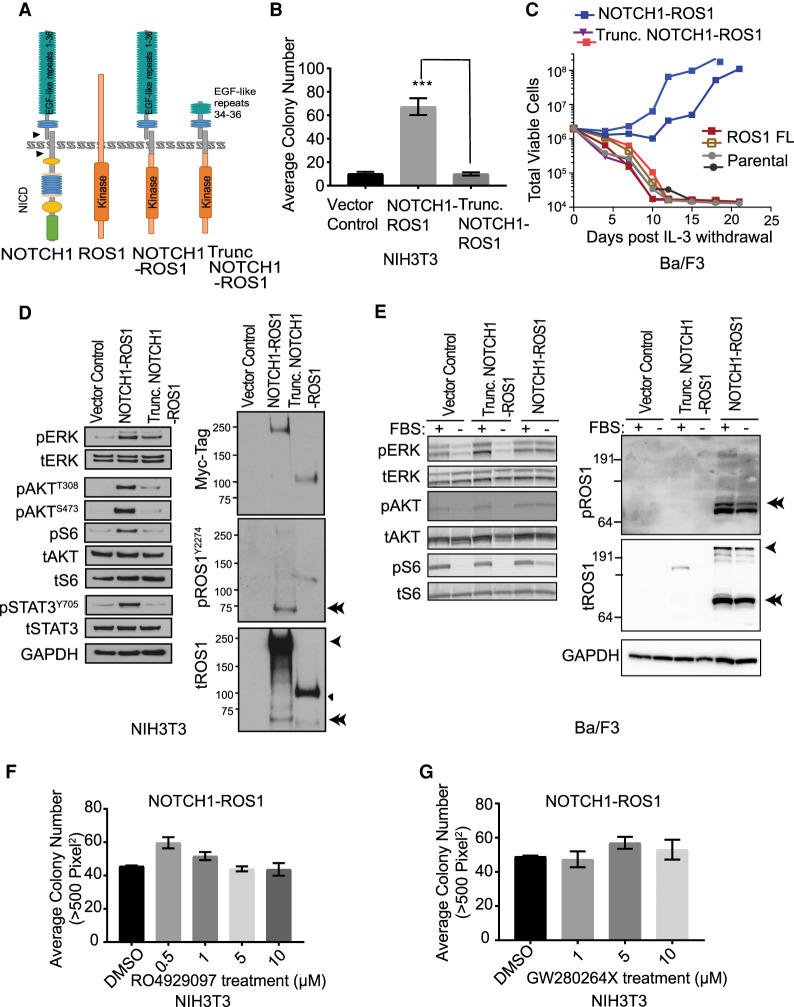
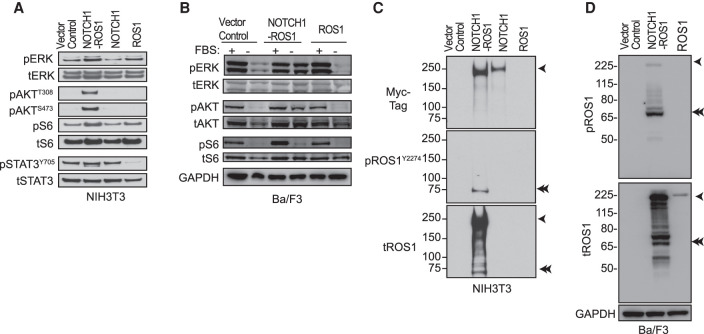
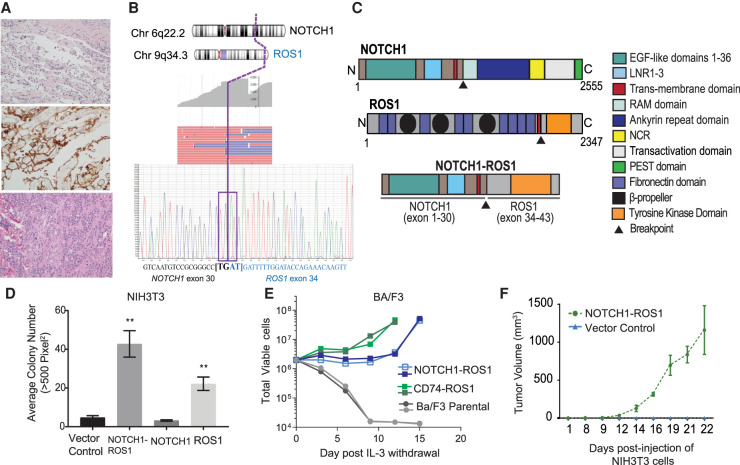
Angiosarcomas are rare, malignant soft tissue tumors in children that arise in a wide range of anatomical locations and have limited targeted therapies available. Here, we report a rare case of a pediatric angiosarcoma (pAS) with Li-Fraumeni syndrome (LFS) expressing a novel NOTCH1-ROS1 gene fusion. Although both NOTCH1 and ROS1 are established proto-oncogenes, our study is the first to describe the mechanistic role of NOTCH1-ROS1 fusion arising via intrachromosomal rearrangement. NOTCH1-ROS1 displayed potent neoplastic transformation propensity in vitro, and harbors tumorigenic potential in vivo, where it induced oncogenic activation of the MAPK, PI3K/mTOR, and JAK-STAT signaling pathways in a murine allograft model. We found an unexpected contribution of the NOTCH1 extracellular region in mediating NOTCH1-ROS1 activation and oncogenic function, highlighting the contribution of both NOTCH1 and ROS1 fusion partners in driving tumorigenicity. Interestingly, neither membrane localization nor fusion protein dimerization were found to be essential for NOTCH1-ROS1 fusion oncogenicity. To target NOTCH1-ROS1-driven tumors, we tested both NOTCH1-directed inhibitors and ROS1-targeted tyrosine kinase inhibitors (TKI) in heterologous models (NIH3T3, Ba/F3). Although NOTCH1 inhibitors did not suppress NOTCH1-ROS1-driven oncogenic growth, we found that oral entrectinib treatment effectively suppressed the growth of NOTCH-ROS1-driven tumors. Taken together, we report the first known pAS case with a novel NOTCH1-ROS1 alteration along with a detailed report on the function and therapeutic targeting of NOTCH1-ROS1. Our study highlights the importance of genomic profiling of rare cancers such as pAS to reveal actionable drivers and improve patient outcomes.
期刊介绍:
Cold Spring Harbor Molecular Case Studies is an open-access, peer-reviewed, international journal in the field of precision medicine. Articles in the journal present genomic and molecular analyses of individuals or cohorts alongside their clinical presentations and phenotypic information. The journal''s purpose is to rapidly share insights into disease development and treatment gained by application of genomics, proteomics, metabolomics, biomarker analysis, and other approaches. The journal covers the fields of cancer, complex diseases, monogenic disorders, neurological conditions, orphan diseases, infectious disease, gene therapy, and pharmacogenomics. It has a rapid peer-review process that is based on technical evaluation of the analyses performed, not the novelty of findings, and offers a swift, clear path to publication. The journal publishes: Research Reports presenting detailed case studies of individuals and small cohorts, Research Articles describing more extensive work using larger cohorts and/or functional analyses, Rapid Communications presenting the discovery of a novel variant and/or novel phenotype associated with a known disease gene, Rapid Cancer Communications presenting the discovery of a novel variant or combination of variants in a cancer type, Variant Discrepancy Resolution describing efforts to resolve differences or update variant interpretations in ClinVar through case-level data sharing, Follow-up Reports linked to previous observations, Plus Review Articles, Editorials, and Position Statements on best practices for research in precision medicine.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
