Elise Vickridge, Camila C F Faraco, Payman S Tehrani, Zubaidah M Ramdzan, Hedyeh Rahimian, Lam Leduy, Anne-Claude Gingras, Alain Nepveu
{"title":"BCL11A的DNA修复功能抑制衰老,促进三阴性乳腺癌细胞的持续增殖。","authors":"Elise Vickridge, Camila C F Faraco, Payman S Tehrani, Zubaidah M Ramdzan, Hedyeh Rahimian, Lam Leduy, Anne-Claude Gingras, Alain Nepveu","doi":"10.1093/narcan/zcac028","DOIUrl":null,"url":null,"abstract":"<p><p>We identified the BCL11A protein in a proximity-dependent biotinylation screen performed with the DNA glycosylase NTHL1. <i>In vitro</i>, DNA repair assays demonstrate that both BCL11A and a small recombinant BCL11A<sup>160-520</sup> protein that is devoid of DNA binding and transcription regulatory domains can stimulate the enzymatic activities of two base excision repair enzymes: NTHL1 and DNA Pol β. Increased DNA repair efficiency, in particular of the base excision repair pathway, is essential for many cancer cells to proliferate in the presence of elevated reactive oxygen species (ROS) produced by cancer-associated metabolic changes. BCL11A is highly expressed in triple-negative breast cancers (TNBC) where its knockdown was reported to reduce clonogenicity and cause tumour regression. We show that <i>BCL11A</i> knockdown in TNBC cells delays repair of oxidative DNA damage, increases the number of oxidized bases and abasic sites in genomic DNA, slows down proliferation and induces cellular senescence. These phenotypes are rescued by ectopic expression of the short BCL11A<sup>160-520</sup> protein. We further show that the BCL11A<sup>160-520</sup> protein accelerates the repair of oxidative DNA damage and cooperates with RAS in cell transformation assays, thereby enabling cells to avoid senescence and continue to proliferate in the presence of high ROS levels.</p>","PeriodicalId":18879,"journal":{"name":"NAR Cancer","volume":"4 4","pages":"zcac028"},"PeriodicalIF":0.0000,"publicationDate":"2022-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/25/a5/zcac028.PMC9516615.pdf","citationCount":"4","resultStr":"{\"title\":\"The DNA repair function of BCL11A suppresses senescence and promotes continued proliferation of triple-negative breast cancer cells.\",\"authors\":\"Elise Vickridge, Camila C F Faraco, Payman S Tehrani, Zubaidah M Ramdzan, Hedyeh Rahimian, Lam Leduy, Anne-Claude Gingras, Alain Nepveu\",\"doi\":\"10.1093/narcan/zcac028\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>We identified the BCL11A protein in a proximity-dependent biotinylation screen performed with the DNA glycosylase NTHL1. <i>In vitro</i>, DNA repair assays demonstrate that both BCL11A and a small recombinant BCL11A<sup>160-520</sup> protein that is devoid of DNA binding and transcription regulatory domains can stimulate the enzymatic activities of two base excision repair enzymes: NTHL1 and DNA Pol β. Increased DNA repair efficiency, in particular of the base excision repair pathway, is essential for many cancer cells to proliferate in the presence of elevated reactive oxygen species (ROS) produced by cancer-associated metabolic changes. BCL11A is highly expressed in triple-negative breast cancers (TNBC) where its knockdown was reported to reduce clonogenicity and cause tumour regression. We show that <i>BCL11A</i> knockdown in TNBC cells delays repair of oxidative DNA damage, increases the number of oxidized bases and abasic sites in genomic DNA, slows down proliferation and induces cellular senescence. These phenotypes are rescued by ectopic expression of the short BCL11A<sup>160-520</sup> protein. We further show that the BCL11A<sup>160-520</sup> protein accelerates the repair of oxidative DNA damage and cooperates with RAS in cell transformation assays, thereby enabling cells to avoid senescence and continue to proliferate in the presence of high ROS levels.</p>\",\"PeriodicalId\":18879,\"journal\":{\"name\":\"NAR Cancer\",\"volume\":\"4 4\",\"pages\":\"zcac028\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2022-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/25/a5/zcac028.PMC9516615.pdf\",\"citationCount\":\"4\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NAR Cancer\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/narcan/zcac028\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NAR Cancer","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/narcan/zcac028","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 4
摘要
我们用DNA糖基化酶NTHL1在邻近依赖的生物素化筛选中鉴定了BCL11A蛋白。体外DNA修复实验表明,BCL11A和缺乏DNA结合和转录调控结构域的小重组BCL11A160-520蛋白都能刺激NTHL1和DNA Pol β两种碱基切除修复酶的酶活性。DNA修复效率的提高,特别是碱基切除修复途径的提高,对于许多癌细胞在癌症相关代谢变化产生的活性氧(ROS)升高的情况下增殖是必不可少的。BCL11A在三阴性乳腺癌(TNBC)中高度表达,据报道其敲低可降低克隆原性并导致肿瘤消退。我们发现,在TNBC细胞中,BCL11A敲低会延迟氧化DNA损伤的修复,增加基因组DNA中氧化碱基和基本位点的数量,减缓增殖并诱导细胞衰老。这些表型通过短BCL11A160-520蛋白的异位表达得以挽救。我们进一步证明BCL11A160-520蛋白加速氧化DNA损伤的修复,并在细胞转化实验中与RAS合作,从而使细胞在高ROS水平下避免衰老并继续增殖。
The DNA repair function of BCL11A suppresses senescence and promotes continued proliferation of triple-negative breast cancer cells.
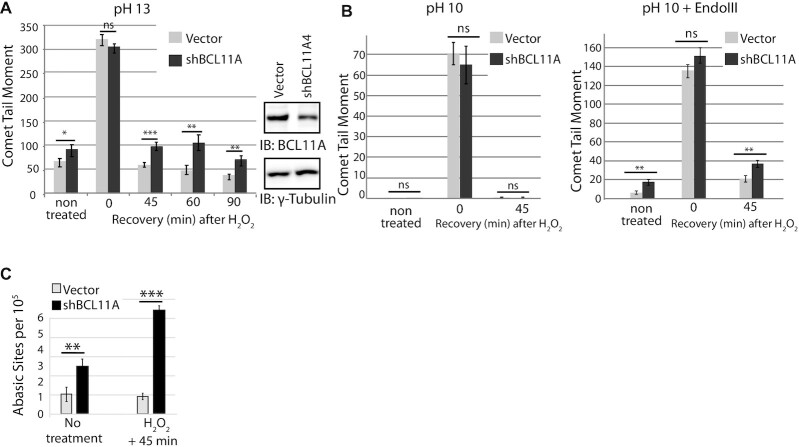
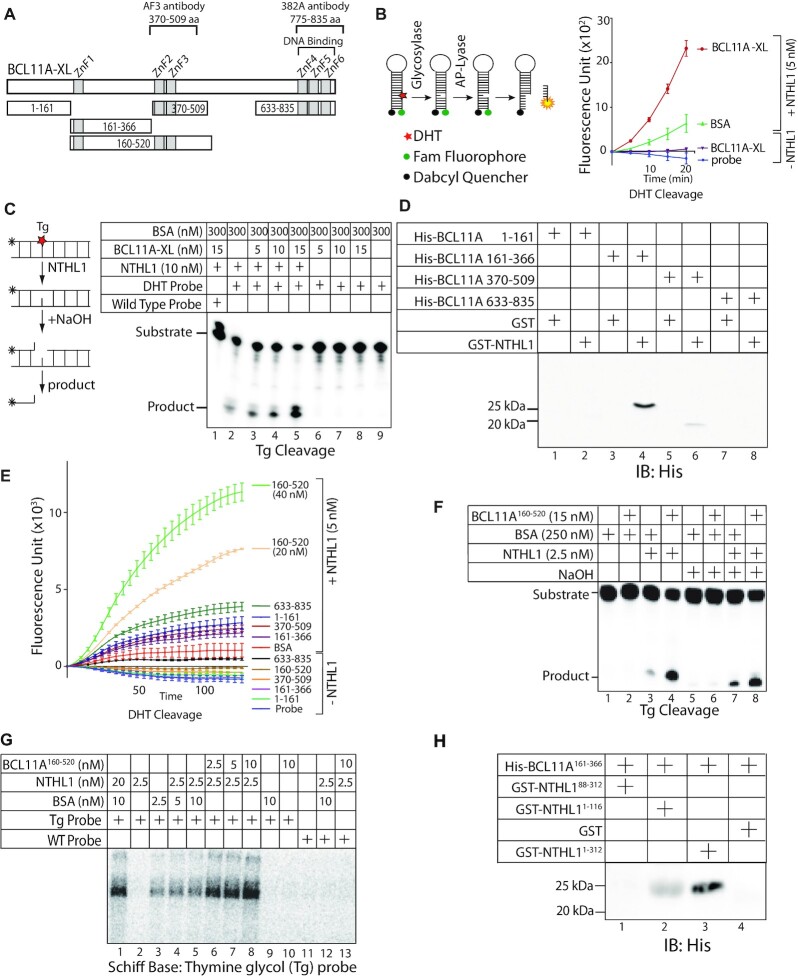
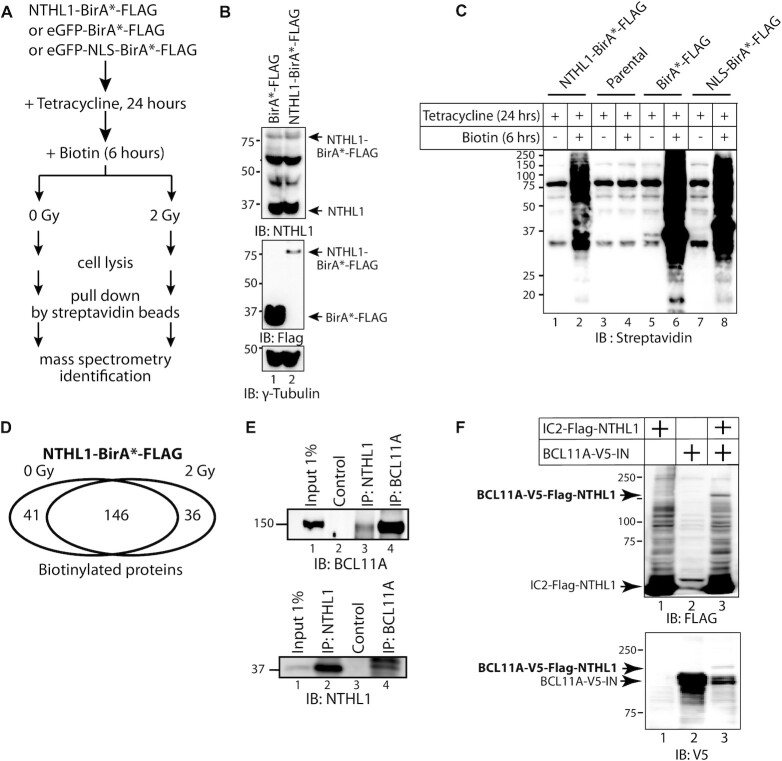
We identified the BCL11A protein in a proximity-dependent biotinylation screen performed with the DNA glycosylase NTHL1. In vitro, DNA repair assays demonstrate that both BCL11A and a small recombinant BCL11A160-520 protein that is devoid of DNA binding and transcription regulatory domains can stimulate the enzymatic activities of two base excision repair enzymes: NTHL1 and DNA Pol β. Increased DNA repair efficiency, in particular of the base excision repair pathway, is essential for many cancer cells to proliferate in the presence of elevated reactive oxygen species (ROS) produced by cancer-associated metabolic changes. BCL11A is highly expressed in triple-negative breast cancers (TNBC) where its knockdown was reported to reduce clonogenicity and cause tumour regression. We show that BCL11A knockdown in TNBC cells delays repair of oxidative DNA damage, increases the number of oxidized bases and abasic sites in genomic DNA, slows down proliferation and induces cellular senescence. These phenotypes are rescued by ectopic expression of the short BCL11A160-520 protein. We further show that the BCL11A160-520 protein accelerates the repair of oxidative DNA damage and cooperates with RAS in cell transformation assays, thereby enabling cells to avoid senescence and continue to proliferate in the presence of high ROS levels.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
