Julie Mouchet, Spyros Roumpanis, Eleni Gaki, Scott Lipnick, Maryam Oskoui, Renata S Scalco, Basil T Darras
{"title":"脊髓性肌萎缩症的疾病负担:使用美国保险索赔数据的比较队列研究。","authors":"Julie Mouchet, Spyros Roumpanis, Eleni Gaki, Scott Lipnick, Maryam Oskoui, Renata S Scalco, Basil T Darras","doi":"10.3233/JND-210764","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Spinal muscular atrophy (SMA) is a neuromuscular disease caused by homozygous deletion or loss-of-function mutations of the survival of motor neuron 1 (SMN1) gene, resulting in reduced levels of SMN protein throughout the body. Patients with SMA may have multiple tissue defects, which could present prior to neuromuscular symptoms.</p><p><strong>Objective: </strong>To assess the signs, comorbidities and potential extraneural manifestations associated with SMA in treatment-naïve patients.</p><p><strong>Methods: </strong>This observational, retrospective and matched-cohort study used secondary insurance claims data from the US IBM® MarketScan® Commercial, Medicaid and Medicare Supplemental databases between 01/01/2000 and 12/31/2013. Treatment-naïve individuals aged≤65 years with≥2 International Classification of Diseases, Ninth Revision (ICD-9) SMA codes were stratified into four groups (A-D), according to age at index (date of first SMA code recorded) and type of ICD-9 code used, and matched with non-SMA controls. The occurrence of ICD-9 codes, which were converted to various classifications (phecodes and system classes), were compared between groups in pre- and post-index periods.</p><p><strong>Results: </strong>A total of 1,457 individuals with SMA were included and matched to 13,362 controls. Increasing numbers of SMA-associated phecodes and system classes were generally observed from pre- to post-index across all groups. The strongest associations were observed in the post-index period for the youngest age groups. Endocrine/metabolic disorders were associated with SMA in almost all groups and across time periods.</p><p><strong>Conclusions: </strong>This exploratory study confirmed the considerable disease burden in patients with SMA and identified 305 unique phecodes associated with SMA, providing a rationale for further research into the natural history and progression of SMA, including extraneural manifestations of the disease.</p>","PeriodicalId":16536,"journal":{"name":"Journal of neuromuscular diseases","volume":"10 1","pages":"41-53"},"PeriodicalIF":3.2000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/b9/f9/jnd-10-jnd210764.PMC9881018.pdf","citationCount":"1","resultStr":"{\"title\":\"Disease Burden of Spinal Muscular Atrophy: A Comparative Cohort Study Using Insurance Claims Data in the USA.\",\"authors\":\"Julie Mouchet, Spyros Roumpanis, Eleni Gaki, Scott Lipnick, Maryam Oskoui, Renata S Scalco, Basil T Darras\",\"doi\":\"10.3233/JND-210764\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Spinal muscular atrophy (SMA) is a neuromuscular disease caused by homozygous deletion or loss-of-function mutations of the survival of motor neuron 1 (SMN1) gene, resulting in reduced levels of SMN protein throughout the body. Patients with SMA may have multiple tissue defects, which could present prior to neuromuscular symptoms.</p><p><strong>Objective: </strong>To assess the signs, comorbidities and potential extraneural manifestations associated with SMA in treatment-naïve patients.</p><p><strong>Methods: </strong>This observational, retrospective and matched-cohort study used secondary insurance claims data from the US IBM® MarketScan® Commercial, Medicaid and Medicare Supplemental databases between 01/01/2000 and 12/31/2013. Treatment-naïve individuals aged≤65 years with≥2 International Classification of Diseases, Ninth Revision (ICD-9) SMA codes were stratified into four groups (A-D), according to age at index (date of first SMA code recorded) and type of ICD-9 code used, and matched with non-SMA controls. The occurrence of ICD-9 codes, which were converted to various classifications (phecodes and system classes), were compared between groups in pre- and post-index periods.</p><p><strong>Results: </strong>A total of 1,457 individuals with SMA were included and matched to 13,362 controls. Increasing numbers of SMA-associated phecodes and system classes were generally observed from pre- to post-index across all groups. The strongest associations were observed in the post-index period for the youngest age groups. Endocrine/metabolic disorders were associated with SMA in almost all groups and across time periods.</p><p><strong>Conclusions: </strong>This exploratory study confirmed the considerable disease burden in patients with SMA and identified 305 unique phecodes associated with SMA, providing a rationale for further research into the natural history and progression of SMA, including extraneural manifestations of the disease.</p>\",\"PeriodicalId\":16536,\"journal\":{\"name\":\"Journal of neuromuscular diseases\",\"volume\":\"10 1\",\"pages\":\"41-53\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/b9/f9/jnd-10-jnd210764.PMC9881018.pdf\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of neuromuscular diseases\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.3233/JND-210764\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of neuromuscular diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.3233/JND-210764","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Disease Burden of Spinal Muscular Atrophy: A Comparative Cohort Study Using Insurance Claims Data in the USA.
Background: Spinal muscular atrophy (SMA) is a neuromuscular disease caused by homozygous deletion or loss-of-function mutations of the survival of motor neuron 1 (SMN1) gene, resulting in reduced levels of SMN protein throughout the body. Patients with SMA may have multiple tissue defects, which could present prior to neuromuscular symptoms.
Objective: To assess the signs, comorbidities and potential extraneural manifestations associated with SMA in treatment-naïve patients.
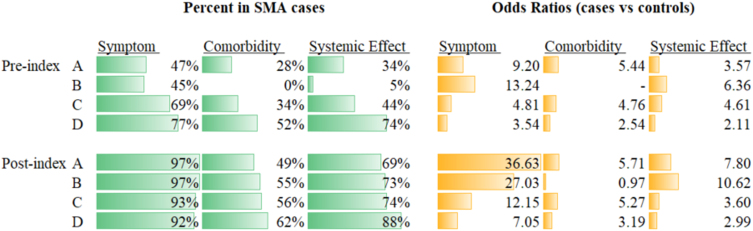
Methods: This observational, retrospective and matched-cohort study used secondary insurance claims data from the US IBM® MarketScan® Commercial, Medicaid and Medicare Supplemental databases between 01/01/2000 and 12/31/2013. Treatment-naïve individuals aged≤65 years with≥2 International Classification of Diseases, Ninth Revision (ICD-9) SMA codes were stratified into four groups (A-D), according to age at index (date of first SMA code recorded) and type of ICD-9 code used, and matched with non-SMA controls. The occurrence of ICD-9 codes, which were converted to various classifications (phecodes and system classes), were compared between groups in pre- and post-index periods.
Results: A total of 1,457 individuals with SMA were included and matched to 13,362 controls. Increasing numbers of SMA-associated phecodes and system classes were generally observed from pre- to post-index across all groups. The strongest associations were observed in the post-index period for the youngest age groups. Endocrine/metabolic disorders were associated with SMA in almost all groups and across time periods.
Conclusions: This exploratory study confirmed the considerable disease burden in patients with SMA and identified 305 unique phecodes associated with SMA, providing a rationale for further research into the natural history and progression of SMA, including extraneural manifestations of the disease.
期刊介绍:
The Journal of Neuromuscular Diseases aims to facilitate progress in understanding the molecular genetics/correlates, pathogenesis, pharmacology, diagnosis and treatment of acquired and genetic neuromuscular diseases (including muscular dystrophy, myasthenia gravis, spinal muscular atrophy, neuropathies, myopathies, myotonias and myositis). The journal publishes research reports, reviews, short communications, letters-to-the-editor, and will consider research that has negative findings. The journal is dedicated to providing an open forum for original research in basic science, translational and clinical research that will improve our fundamental understanding and lead to effective treatments of neuromuscular diseases.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
