Jong-Lyul Park, Jae-Yoon Kim, Seon-Young Kim, Yong Sun Lee
{"title":"人乳腺上皮细胞全基因组测序数据的生成与分析。","authors":"Jong-Lyul Park, Jae-Yoon Kim, Seon-Young Kim, Yong Sun Lee","doi":"10.5808/gi.22044","DOIUrl":null,"url":null,"abstract":"<p><p>Breast cancer is the most common cancer worldwide, and advanced breast cancer with metastases is incurable mainly with currently available therapies. Therefore, it is essential to understand molecular characteristics during the progression of breast carcinogenesis. Here, we report a dataset of whole genomes from the human mammary epithelial cell system derived from a reduction mammoplasty specimen. This system comprises pre-stasis 184D cells, considered normal, and seven cell lines along cancer progression series that are immortalized or additionally acquired anchorage-independent growth. Our analysis of the whole-genome sequencing (WGS) data indicates that those seven cancer progression series cells have somatic mutations whose number ranges from 8,393 to 39,564 (with an average of 30,591) compared to 184D cells. These WGS data and our mutation analysis will provide helpful information to identify driver mutations and elucidate molecular mechanisms for breast carcinogenesis.</p>","PeriodicalId":36591,"journal":{"name":"Genomics and Informatics","volume":"21 1","pages":"e11"},"PeriodicalIF":0.0000,"publicationDate":"2023-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10085740/pdf/","citationCount":"0","resultStr":"{\"title\":\"Generation and analysis of whole-genome sequencing data in human mammary epithelial cells.\",\"authors\":\"Jong-Lyul Park, Jae-Yoon Kim, Seon-Young Kim, Yong Sun Lee\",\"doi\":\"10.5808/gi.22044\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Breast cancer is the most common cancer worldwide, and advanced breast cancer with metastases is incurable mainly with currently available therapies. Therefore, it is essential to understand molecular characteristics during the progression of breast carcinogenesis. Here, we report a dataset of whole genomes from the human mammary epithelial cell system derived from a reduction mammoplasty specimen. This system comprises pre-stasis 184D cells, considered normal, and seven cell lines along cancer progression series that are immortalized or additionally acquired anchorage-independent growth. Our analysis of the whole-genome sequencing (WGS) data indicates that those seven cancer progression series cells have somatic mutations whose number ranges from 8,393 to 39,564 (with an average of 30,591) compared to 184D cells. These WGS data and our mutation analysis will provide helpful information to identify driver mutations and elucidate molecular mechanisms for breast carcinogenesis.</p>\",\"PeriodicalId\":36591,\"journal\":{\"name\":\"Genomics and Informatics\",\"volume\":\"21 1\",\"pages\":\"e11\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-03-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10085740/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Genomics and Informatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.5808/gi.22044\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"Agricultural and Biological Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genomics and Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5808/gi.22044","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
Generation and analysis of whole-genome sequencing data in human mammary epithelial cells.
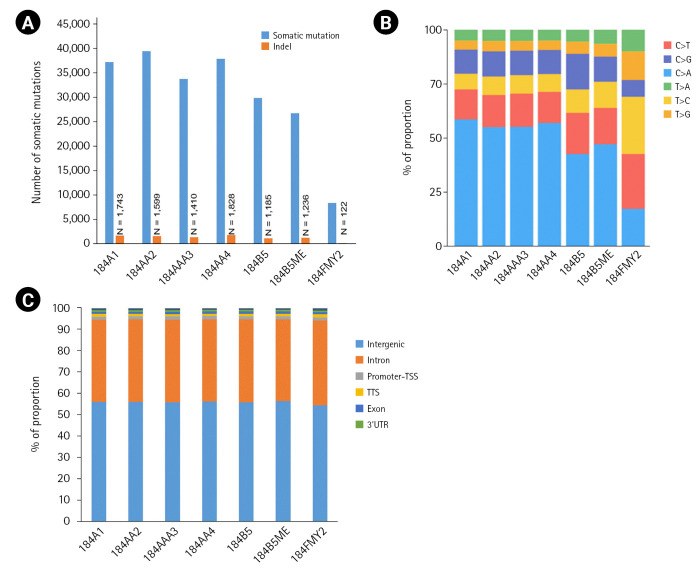
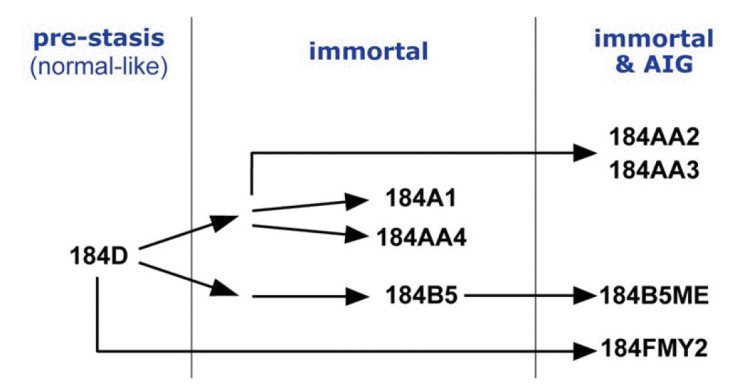
Breast cancer is the most common cancer worldwide, and advanced breast cancer with metastases is incurable mainly with currently available therapies. Therefore, it is essential to understand molecular characteristics during the progression of breast carcinogenesis. Here, we report a dataset of whole genomes from the human mammary epithelial cell system derived from a reduction mammoplasty specimen. This system comprises pre-stasis 184D cells, considered normal, and seven cell lines along cancer progression series that are immortalized or additionally acquired anchorage-independent growth. Our analysis of the whole-genome sequencing (WGS) data indicates that those seven cancer progression series cells have somatic mutations whose number ranges from 8,393 to 39,564 (with an average of 30,591) compared to 184D cells. These WGS data and our mutation analysis will provide helpful information to identify driver mutations and elucidate molecular mechanisms for breast carcinogenesis.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
