Emma Bigelow, Suchi Saria, Brian Piening, Brendan Curti, Alexa Dowdell, Roshanthi Weerasinghe, Carlo Bifulco, Walter Urba, Noam Finkelstein, Elana J Fertig, Alex Baras, Neeha Zaidi, Elizabeth Jaffee, Mark Yarchoan
{"title":"用于抗 PD1 免疫疗法反应的肿瘤不可知性预测的随机森林基因组分类器。","authors":"Emma Bigelow, Suchi Saria, Brian Piening, Brendan Curti, Alexa Dowdell, Roshanthi Weerasinghe, Carlo Bifulco, Walter Urba, Noam Finkelstein, Elana J Fertig, Alex Baras, Neeha Zaidi, Elizabeth Jaffee, Mark Yarchoan","doi":"10.1177/11769351221136081","DOIUrl":null,"url":null,"abstract":"<p><p>Tumor mutational burden (TMB), a surrogate for tumor neoepitope burden, is used as a pan-tumor biomarker to identify patients who may benefit from anti-program cell death 1 (PD1) immunotherapy, but it is an imperfect biomarker. Multiple additional genomic characteristics are associated with anti-PD1 responses, but the combined predictive value of these features and the added informativeness of each respective feature remains unknown. We evaluated whether machine learning (ML) approaches using proposed determinants of anti-PD1 response derived from whole exome sequencing (WES) could improve prediction of anti-PD1 responders over TMB alone. Random forest classifiers were trained on publicly available anti-PD1 data (n = 104), and subsequently tested on an independent anti-PD1 cohort (n = 69). Both the training and test datasets included a range of cancer types such as non-small cell lung cancer (NSCLC), head and neck squamous cell carcinoma (HNSCC), melanoma, and smaller numbers of patients from other tumor types. Features used include summaries such as TMB and number of frameshift mutations, as well as more gene-level features such as counts of mutations associated with immune checkpoint response and resistance. Both ML algorithms demonstrated area under the receiver-operator curves (AUC) that exceeded TMB alone (AUC 0.63 \"human-guided,\" 0.64 \"cluster,\" and 0.58 TMB alone). Mutations within oncogenes disproportionately modulate anti-PD1 responses relative to their overall contribution to tumor neoepitope burden. The use of a ML algorithm evaluating multiple proposed genomic determinants of anti-PD1 responses modestly improves performance over TMB alone, highlighting the need to integrate other biomarkers to further improve model performance.</p>","PeriodicalId":35418,"journal":{"name":"Cancer Informatics","volume":"21 ","pages":"11769351221136081"},"PeriodicalIF":2.1000,"publicationDate":"2022-11-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/c7/0c/10.1177_11769351221136081.PMC9685115.pdf","citationCount":"0","resultStr":"{\"title\":\"A Random Forest Genomic Classifier for Tumor Agnostic Prediction of Response to Anti-PD1 Immunotherapy.\",\"authors\":\"Emma Bigelow, Suchi Saria, Brian Piening, Brendan Curti, Alexa Dowdell, Roshanthi Weerasinghe, Carlo Bifulco, Walter Urba, Noam Finkelstein, Elana J Fertig, Alex Baras, Neeha Zaidi, Elizabeth Jaffee, Mark Yarchoan\",\"doi\":\"10.1177/11769351221136081\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Tumor mutational burden (TMB), a surrogate for tumor neoepitope burden, is used as a pan-tumor biomarker to identify patients who may benefit from anti-program cell death 1 (PD1) immunotherapy, but it is an imperfect biomarker. Multiple additional genomic characteristics are associated with anti-PD1 responses, but the combined predictive value of these features and the added informativeness of each respective feature remains unknown. We evaluated whether machine learning (ML) approaches using proposed determinants of anti-PD1 response derived from whole exome sequencing (WES) could improve prediction of anti-PD1 responders over TMB alone. Random forest classifiers were trained on publicly available anti-PD1 data (n = 104), and subsequently tested on an independent anti-PD1 cohort (n = 69). Both the training and test datasets included a range of cancer types such as non-small cell lung cancer (NSCLC), head and neck squamous cell carcinoma (HNSCC), melanoma, and smaller numbers of patients from other tumor types. Features used include summaries such as TMB and number of frameshift mutations, as well as more gene-level features such as counts of mutations associated with immune checkpoint response and resistance. Both ML algorithms demonstrated area under the receiver-operator curves (AUC) that exceeded TMB alone (AUC 0.63 \\\"human-guided,\\\" 0.64 \\\"cluster,\\\" and 0.58 TMB alone). Mutations within oncogenes disproportionately modulate anti-PD1 responses relative to their overall contribution to tumor neoepitope burden. The use of a ML algorithm evaluating multiple proposed genomic determinants of anti-PD1 responses modestly improves performance over TMB alone, highlighting the need to integrate other biomarkers to further improve model performance.</p>\",\"PeriodicalId\":35418,\"journal\":{\"name\":\"Cancer Informatics\",\"volume\":\"21 \",\"pages\":\"11769351221136081\"},\"PeriodicalIF\":2.1000,\"publicationDate\":\"2022-11-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/c7/0c/10.1177_11769351221136081.PMC9685115.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cancer Informatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/11769351221136081\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2022/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cancer Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/11769351221136081","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
A Random Forest Genomic Classifier for Tumor Agnostic Prediction of Response to Anti-PD1 Immunotherapy.
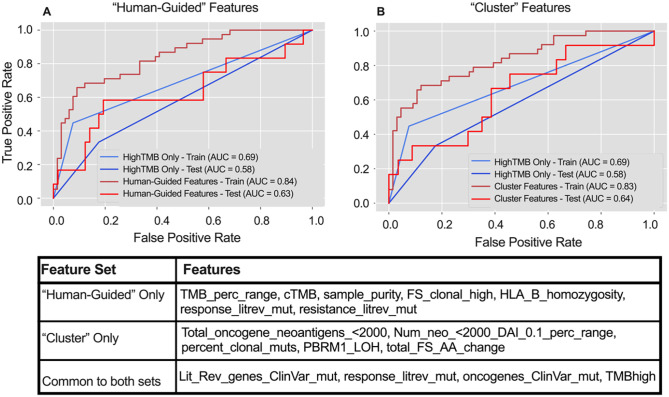
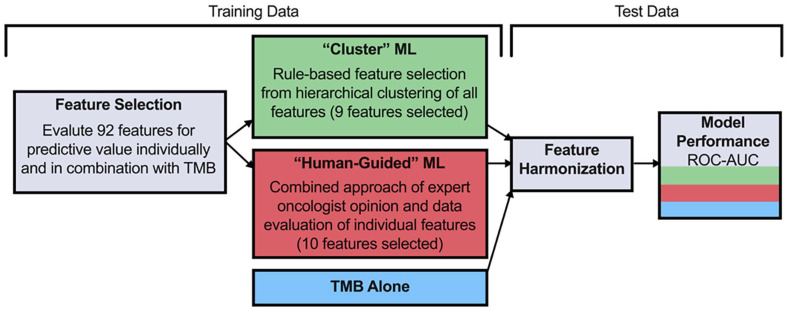
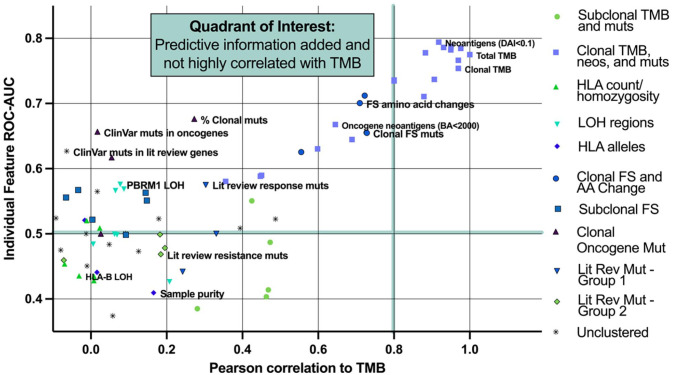
Tumor mutational burden (TMB), a surrogate for tumor neoepitope burden, is used as a pan-tumor biomarker to identify patients who may benefit from anti-program cell death 1 (PD1) immunotherapy, but it is an imperfect biomarker. Multiple additional genomic characteristics are associated with anti-PD1 responses, but the combined predictive value of these features and the added informativeness of each respective feature remains unknown. We evaluated whether machine learning (ML) approaches using proposed determinants of anti-PD1 response derived from whole exome sequencing (WES) could improve prediction of anti-PD1 responders over TMB alone. Random forest classifiers were trained on publicly available anti-PD1 data (n = 104), and subsequently tested on an independent anti-PD1 cohort (n = 69). Both the training and test datasets included a range of cancer types such as non-small cell lung cancer (NSCLC), head and neck squamous cell carcinoma (HNSCC), melanoma, and smaller numbers of patients from other tumor types. Features used include summaries such as TMB and number of frameshift mutations, as well as more gene-level features such as counts of mutations associated with immune checkpoint response and resistance. Both ML algorithms demonstrated area under the receiver-operator curves (AUC) that exceeded TMB alone (AUC 0.63 "human-guided," 0.64 "cluster," and 0.58 TMB alone). Mutations within oncogenes disproportionately modulate anti-PD1 responses relative to their overall contribution to tumor neoepitope burden. The use of a ML algorithm evaluating multiple proposed genomic determinants of anti-PD1 responses modestly improves performance over TMB alone, highlighting the need to integrate other biomarkers to further improve model performance.
期刊介绍:
The field of cancer research relies on advances in many other disciplines, including omics technology, mass spectrometry, radio imaging, computer science, and biostatistics. Cancer Informatics provides open access to peer-reviewed high-quality manuscripts reporting bioinformatics analysis of molecular genetics and/or clinical data pertaining to cancer, emphasizing the use of machine learning, artificial intelligence, statistical algorithms, advanced imaging techniques, data visualization, and high-throughput technologies. As the leading journal dedicated exclusively to the report of the use of computational methods in cancer research and practice, Cancer Informatics leverages methodological improvements in systems biology, genomics, proteomics, metabolomics, and molecular biochemistry into the fields of cancer detection, treatment, classification, risk-prediction, prevention, outcome, and modeling.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
