Nourhan Abu-Shahba, Elsayed Hegazy, Faiz M Khan, Mahmoud Elhefnawi
{"title":"肝癌组织MicroRNA表达数据的计算机分析。","authors":"Nourhan Abu-Shahba, Elsayed Hegazy, Faiz M Khan, Mahmoud Elhefnawi","doi":"10.1177/11769351231171743","DOIUrl":null,"url":null,"abstract":"<p><p>Abnormal miRNA expression has been evidenced to be directly linked to HCC initiation and progression. This study was designed to detect possible prognostic, diagnostic, and/or therapeutic miRNAs for HCC using computational analysis of miRNAs expression. Methods: miRNA expression datasets meta-analysis was performed using the YM500v2 server to compare miRNA expression in normal and cancerous liver tissues. The most significant differentially regulated miRNAs in our study undergone target gene analysis using the mirWalk tool to obtain their validated and predicted targets. The combinatorial target prediction tool; miRror Suite was used to obtain the commonly regulated target genes. Functional enrichment analysis was performed on the resulting targets using the DAVID tool. A network was constructed based on interactions among microRNAs, their targets, and transcription factors. Hub nodes and gatekeepers were identified using network topological analysis. Further, we performed patient data survival analysis based on low and high expression of identified hubs and gatekeeper nodes, patients were stratified into low and high survival probability groups. Results: Using the meta-analysis option in the YM500v2 server, 34 miRNAs were found to be significantly differentially regulated (<i>P</i>-value ⩽ .05); 5 miRNAs were down-regulated while 29 were up-regulated. The validated and predicted target genes for each miRNA, as well as the combinatorially predicted targets, were obtained. DAVID enrichment analysis resulted in several important cellular functions that are directly related to the main cancer hallmarks. Among these functions are focal adhesion, cell cycle, PI3K-Akt signaling, insulin signaling, Ras and MAPK signaling pathways. Several hub genes and gatekeepers were found that could serve as potential drug targets for hepatocellular carcinoma. POU2F1 and PPARA showed a significant difference between low and high survival probabilities (<i>P</i>-value ⩽ .05) in HCC patients. Our study sheds light on important biomarker miRNAs for hepatocellular carcinoma along with their target genes and their regulated functions.</p>","PeriodicalId":35418,"journal":{"name":"Cancer Informatics","volume":"22 ","pages":"11769351231171743"},"PeriodicalIF":2.5000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/64/09/10.1177_11769351231171743.PMC10185868.pdf","citationCount":"0","resultStr":"{\"title\":\"In Silico Analysis of MicroRNA Expression Data in Liver Cancer.\",\"authors\":\"Nourhan Abu-Shahba, Elsayed Hegazy, Faiz M Khan, Mahmoud Elhefnawi\",\"doi\":\"10.1177/11769351231171743\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Abnormal miRNA expression has been evidenced to be directly linked to HCC initiation and progression. This study was designed to detect possible prognostic, diagnostic, and/or therapeutic miRNAs for HCC using computational analysis of miRNAs expression. Methods: miRNA expression datasets meta-analysis was performed using the YM500v2 server to compare miRNA expression in normal and cancerous liver tissues. The most significant differentially regulated miRNAs in our study undergone target gene analysis using the mirWalk tool to obtain their validated and predicted targets. The combinatorial target prediction tool; miRror Suite was used to obtain the commonly regulated target genes. Functional enrichment analysis was performed on the resulting targets using the DAVID tool. A network was constructed based on interactions among microRNAs, their targets, and transcription factors. Hub nodes and gatekeepers were identified using network topological analysis. Further, we performed patient data survival analysis based on low and high expression of identified hubs and gatekeeper nodes, patients were stratified into low and high survival probability groups. Results: Using the meta-analysis option in the YM500v2 server, 34 miRNAs were found to be significantly differentially regulated (<i>P</i>-value ⩽ .05); 5 miRNAs were down-regulated while 29 were up-regulated. The validated and predicted target genes for each miRNA, as well as the combinatorially predicted targets, were obtained. DAVID enrichment analysis resulted in several important cellular functions that are directly related to the main cancer hallmarks. Among these functions are focal adhesion, cell cycle, PI3K-Akt signaling, insulin signaling, Ras and MAPK signaling pathways. Several hub genes and gatekeepers were found that could serve as potential drug targets for hepatocellular carcinoma. POU2F1 and PPARA showed a significant difference between low and high survival probabilities (<i>P</i>-value ⩽ .05) in HCC patients. Our study sheds light on important biomarker miRNAs for hepatocellular carcinoma along with their target genes and their regulated functions.</p>\",\"PeriodicalId\":35418,\"journal\":{\"name\":\"Cancer Informatics\",\"volume\":\"22 \",\"pages\":\"11769351231171743\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/64/09/10.1177_11769351231171743.PMC10185868.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cancer Informatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/11769351231171743\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cancer Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/11769351231171743","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
In Silico Analysis of MicroRNA Expression Data in Liver Cancer.
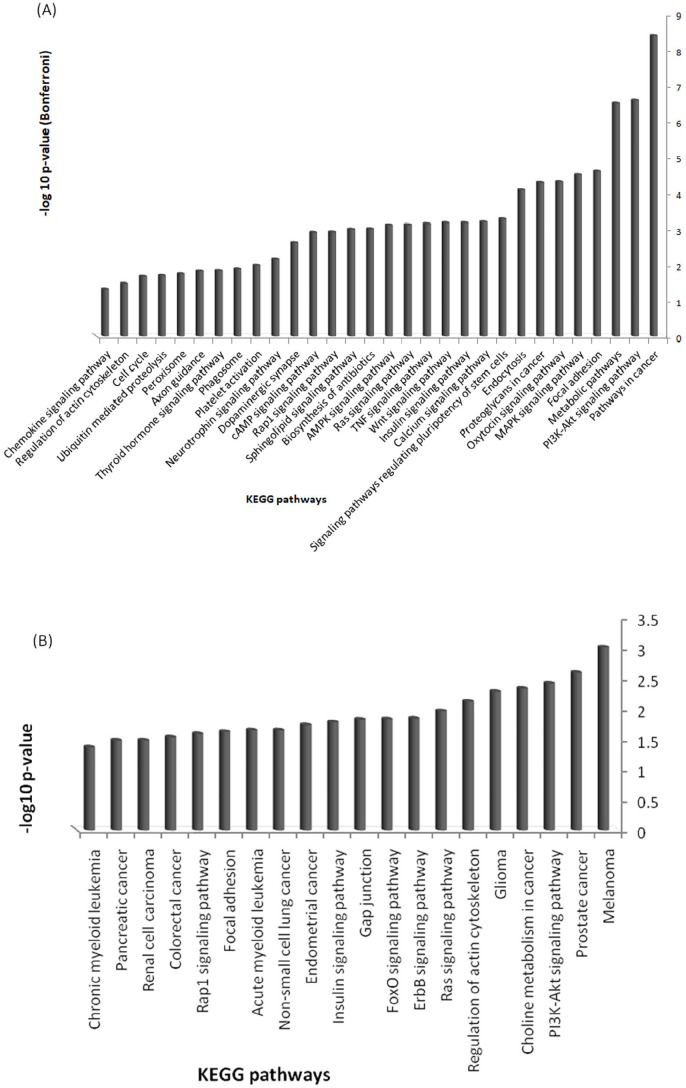
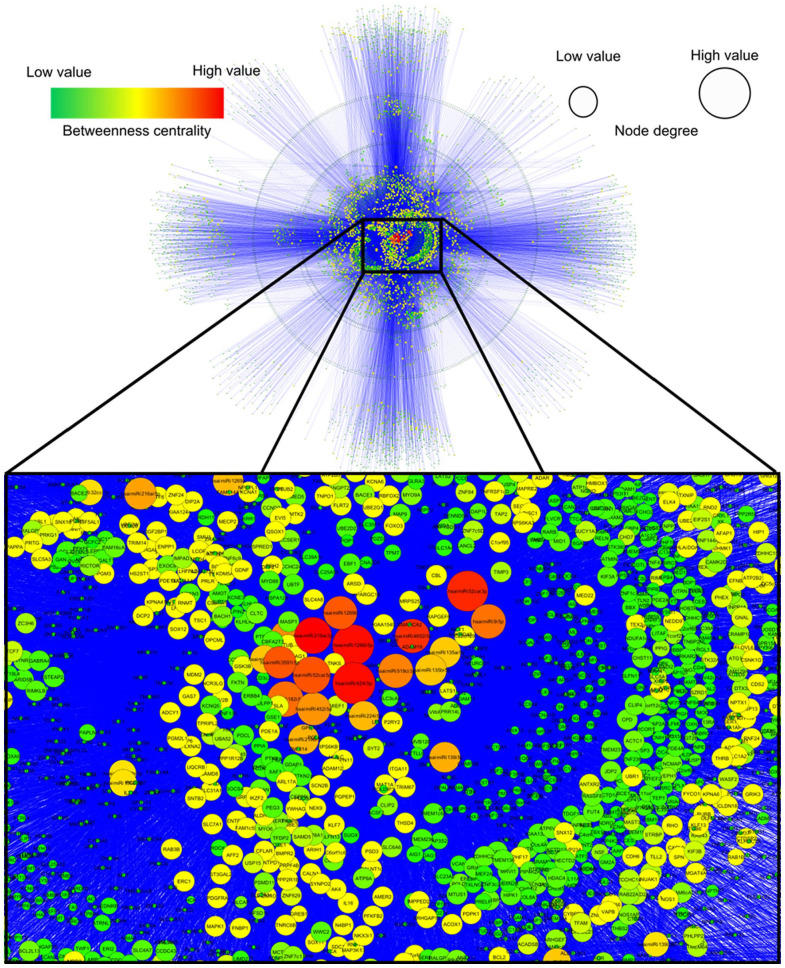
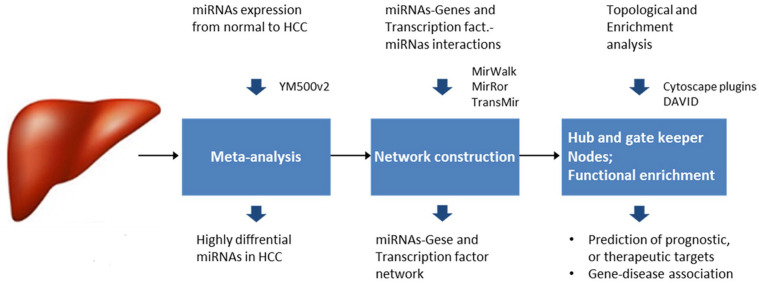
Abnormal miRNA expression has been evidenced to be directly linked to HCC initiation and progression. This study was designed to detect possible prognostic, diagnostic, and/or therapeutic miRNAs for HCC using computational analysis of miRNAs expression. Methods: miRNA expression datasets meta-analysis was performed using the YM500v2 server to compare miRNA expression in normal and cancerous liver tissues. The most significant differentially regulated miRNAs in our study undergone target gene analysis using the mirWalk tool to obtain their validated and predicted targets. The combinatorial target prediction tool; miRror Suite was used to obtain the commonly regulated target genes. Functional enrichment analysis was performed on the resulting targets using the DAVID tool. A network was constructed based on interactions among microRNAs, their targets, and transcription factors. Hub nodes and gatekeepers were identified using network topological analysis. Further, we performed patient data survival analysis based on low and high expression of identified hubs and gatekeeper nodes, patients were stratified into low and high survival probability groups. Results: Using the meta-analysis option in the YM500v2 server, 34 miRNAs were found to be significantly differentially regulated (P-value ⩽ .05); 5 miRNAs were down-regulated while 29 were up-regulated. The validated and predicted target genes for each miRNA, as well as the combinatorially predicted targets, were obtained. DAVID enrichment analysis resulted in several important cellular functions that are directly related to the main cancer hallmarks. Among these functions are focal adhesion, cell cycle, PI3K-Akt signaling, insulin signaling, Ras and MAPK signaling pathways. Several hub genes and gatekeepers were found that could serve as potential drug targets for hepatocellular carcinoma. POU2F1 and PPARA showed a significant difference between low and high survival probabilities (P-value ⩽ .05) in HCC patients. Our study sheds light on important biomarker miRNAs for hepatocellular carcinoma along with their target genes and their regulated functions.
期刊介绍:
The field of cancer research relies on advances in many other disciplines, including omics technology, mass spectrometry, radio imaging, computer science, and biostatistics. Cancer Informatics provides open access to peer-reviewed high-quality manuscripts reporting bioinformatics analysis of molecular genetics and/or clinical data pertaining to cancer, emphasizing the use of machine learning, artificial intelligence, statistical algorithms, advanced imaging techniques, data visualization, and high-throughput technologies. As the leading journal dedicated exclusively to the report of the use of computational methods in cancer research and practice, Cancer Informatics leverages methodological improvements in systems biology, genomics, proteomics, metabolomics, and molecular biochemistry into the fields of cancer detection, treatment, classification, risk-prediction, prevention, outcome, and modeling.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
